 ExPASy Home page ExPASy Home page |
Site Map | Search ExPASy | Contact us | Proteomics tools |
| ExPASy Home page |
Site Map | Search ExPASy | Contact us | Proteomics tools |
| Hosted by NCSC US | Mirror sites: | Canada | China | Korea | Switzerland | Taiwan |
Pour voir la documentation technique clicker ici.
Ce projet est une extension d'une application bio-informatique PeptIdent (voir ci-dessous) et permet la visualisation sous forme graphique des résultats de ce dernier obtenus avec des données specifiées par l'utilisateur. Les notions de base en biochimie suivantes sont utilisées par BioGraph, le logiciel que nous avons developpé:
- Proteine est une substance formée de chaines d'acides aminés (20) qui sont attachés par des peptide bonds.
- Peptide est une molecule qui comprend deux ou plus molecules d'acide aminé.
- Acide aminé est une molecule principalement composée de carbone, oxygène, hydrogène et nitrogène.
- Missed clevage est un terme employé pour signifier que la proteine c'est mal divisé en peptides (il manque une ou plusieurs divisions).
Une proteine peut avoir des ponts de sulfure, c'est à dire que deux acides aminés qui ne sont pas voisins directs forme un pont entre eux, ainsi que des modifications post-traductionnelles càd qu'il y a, entre autres, des sucres qui viennent s'ajouter a cette proteine.
PeptIdent est un outil qui permet l'identification de proteines à partir des masses expérimentalement obtenues (avec un spectromètre de masse), comparées avec des valeurs théoriques de peptides calculées pour toutes les protéines se trouvant dans la base de données SWISS-PROT. Ces masses expérimentales sont soit fournies par l'utilisateur telles quelles, soit par le résultat d'une digéstion par un spectrometre de masse dans un format PKM:
"Peak Table"
OP=0
Center X Peak Y Left X Right X Time X Mass Difference Name
STD.Misc Height Left Y Right Y %Height,Width,%Area,%Quan,H/A
930.67310 17719.000 930.56730 930.78710 49127.310 0 930.67
C 0.? 0 12403.000 12403.000
1027.8450 53725.000 1027.7270 1027.9600 51622.450 0 1027.85
C 0.? 0 37608.000 37608.000
1141.9430 8096.0000 1141.6660 1142.2410 54406.030 0 1141.94
C 0.? 0 5667.0000 5667.0000
1193.0070 8396.0000 1193.0070 1193.0070 55606.580 0 1193.01
C 0.? 0 8396.0000 8396.0000
1203.8100 9386.0000 1203.6650 1203.9510 55857.260 0 1203.81
C 0.? 0 6570.0000 6570.0000
1214.9160 11820.000 1214.7920 1215.0460 56113.810 0 1214.92
C 0.? 0 8274.0000 8274.0000
1233.9170 59641.000 1233.7840 1234.0470 56549.990 0 1233.92
C 0.? 0 41749.000 41749.000
1247.8670 3789.0000 1247.5570 1248.1200 56868.120 0 1247.87
C 0.? 0 2652.0000 2652.0000
1305.876 5366 1305.876 1305.876 58172.24 0 1305.88
C 0.? 0 5366 5366
1390.9880 9860.0000 1390.8450 1391.1320 60034.330 0 1390.99
C 0.? 0 6902.0000 6902.0000
1446.148 12203 1446.011 1446.281 61210.82 0 1446.15
C 0.? 0 8542 8542
1490.226 5531 1490.059 1490.393 62134.92 0 1490.23
C 0.? 0 3872 3872
1618.171 23431 1618.022 1618.319 64742.47 0 1618.17
C 0.? 0 16402 16402
1632.1910 44844.000 1632.0510 1632.3350 65021.830 0 1632.19
C 0.? 0 31391.000 31391.000
1751.19 10639 1750.904 1751.492 67346.28 0 1751.19
C 0.? 0 7447 7447
1796.399 10456 1796.224 1796.583 68208.57 0 1796.40
C 0.? 0 7319 7319
1804.3260 40386.000 1804.1660 1804.4860 68358.660 0 1804.33
C 0.? 0 28270.000 28270.000
1813.372 10006 1813.178 1813.559 68529.51 0 1813.37
C 0.? 0 7004 7004
1962.5200 51900.000 1962.3420 1962.6950 71287.410 0 1962.52
C 0.? 0 36330.000 36330.000
1965.417 53714 1965.417 1965.417 71339.91 0 1965.42
C 0.? 0 53714 53714
2117.6890 25456.000 2117.5150 2117.8640 74047.530 0 2117.69
C 0.? 0 17819.000 17819.000Fig.1 : Fichier PKM (0609_15.pkm)
PeptIdent fournit une série de proteines et des peptides qui les constituent sous forme d'un simple tableau. Le nombre de résulats peut croître d'une manière exponentielle selon les critères choisis au lancement de ce dernier. On constate, bien que les résultats soient sous la forme de tableaux bien organisés et clairs, qu'il n'est pas forcement évident de retrouver la bonne proteine dans une serie de chiffres plus ou moins significatifs. Voici un résultat de PeptIdent sur un fichier PKM (on a affiché que la première proteine correspondante)
| Name given to unknown protein: | unknown | ||||
| Species searched: | all | ||||
pI: 0 -  |
|||||
| Mw: 0 - |
|||||
| Peptide masses for unknown protein: | 930.67310 1027.8450 1141.9430 1193.0070 1203.8100 1214.9160 1233.9170 1247.8670 1305.876 1390.9880 1446.148 1490.226 1618.171 1632.1910 1751.19 1796.399 1804.3260 1813.372 1962.5200 1965.417 2117.6890 | ||||
| Tolerance: | ±0.5 Dalton | ||||
| Minimum number of peptides required to match: | 4 | ||||
| Maximum number of matching proteins to print: | 50 | ||||
| Using monoisotopic masses of the occurring amino acid residues and interpreting your peptide masses as [M+H]+. | |||||
| Enzyme: Trypsin, allowing for up to 0 missed cleavages (#MC). | |||||
| Cysteine in reduced form. | |||||
| Scan done on 14-Nov-1998, SWISS-PROT Release 36 and updates up to 07-Nov-1998: 76502 entries . | |||||
| Score | # peptide matches | AC | ID | Description | pI | Mw |
|---|---|---|---|---|---|---|
| 0.43 | 9 | P21694 | EFTU_SALTY | ELONGATION FACTOR TU (EF-TU). | 5.30 | 43152.36 |
| 0.43 | 9 | P02990 | EFTU_ECOLI | ELONGATION FACTOR TU (EF-TU) (P-43). | 5.30 | 43182.39 |
| 0.29 | 6 | P97929 | BRC2_MOUSE | BREAST CANCER TYPE 2 SUSCEPTIBILITY PROTEIN. | 6.23 | 370663.75 |
| 0.29 | 6 | P27743 | ACVS_NOCLA | DELTA-(L-ALPHA-AMINOADIPYL)-L-CYSTEINYL-D-VALINE SYNTHETASE (EC 6.-.-.-) (ACV SYNTHETASE) (ACVS). | 5.21 | 404087.83 |
| 0.29 | 6 | P45444 | DYHC_EMENI | DYNEIN HEAVY CHAIN, CYTOSOLIC (DYHC). | 5.72 | 492480.49 |
| 0.24 | 5 | P23098 | DYHC_TRIGR | DYNEIN BETA CHAIN, CILIARY. | 5.22 | 511782.00 |
| 0.24 | 5 | P55037 | GLTB_SYNY3 | FERREDOXIN-DEPENDENT GLUTAMATE SYNTHASE 1 (EC 1.4.7.1) (FD-GOGAT). | 5.60 | 169071.50 |
| 0.24 | 5 | P49792 | N358_HUMAN | NUCLEAR PORE COMPLEX PROTEIN NUP358 (NUCLEOPORIN NUP358) (358 KD NUCLEOPORIN) (P270) (RAN-BINDING PROTEIN 2). | 5.86 | 358218.28 |
| 0.24 | 5 | P12263 | FA8_PIG_1 | CHAIN 1: COAGULATION FACTOR VIII. | 6.53 | 237208.69 |
| 0.24 | 5 | O13710 | YDZ2_SCHPO | HYPOTHETICAL 123.7 KD PROTEIN C14C4.02 IN CHROMOSOME I. | 5.83 | 123722.68 |
| 0.24 | 5 | Q02224 | CENE_HUMAN | CENTROMERIC PROTEIN E (CENP-E PROTEIN). | 5.46 | 312090.24 |
| 0.24 | 5 | Q05057 | POLG_PYFV1 | GENOME POLYPROTEIN [CONTAINS: 22.5 KD PROTEIN; 26 KD PROTEIN; 31 KD PROTEIN; PROBABLE RNA-DIRECTED RNA POLYMERASE (EC 2.7.7.48)]. | 6.57 | 336247.40 |
| 0.24 | 5 | Q01886 | HTS1_COCCA | HC-TOXIN SYNTHETASE (EC 6.3.2.-) (HTS). | 5.80 | 574651.99 |
| 0.24 | 5 | P43926 | EFTU_HAEIN | ELONGATION FACTOR TU (EF-TU). | 5.26 | 43223.17 |
| 0.24 | 5 | Q05793 | PGBM_MOUSE_1 | CHAIN 1: BASEMENT MEMBRANE-SPECIFIC HEPARAN | 5.85 | 396109.26 |
| 0.24 | 5 | P33053 | RPO1_VARV | DNA-DIRECTED RNA POLYMERASE 147 KD POLYPEPTIDE (EC 2.7.7.6). | 8.10 | 146783.34 |
| 0.19 | 4 | P53125 | YGN3_YEAST | HYPOTHETICAL 145.6 KD PROTEIN IN SSM1B-CEG1 INTERGENIC REGION. | 5.50 | 145642.91 |
| 0.19 | 4 | P44781 | HEPA_HAEIN | RNA POLYMERASE ASSOCIATED PROTEIN HOMOLOG (ATP-DEPENDENT HELICASE HEPA). | 5.32 | 104405.28 |
| 0.19 | 4 | P20054 | PYR1_DICDI | PROTEIN PYR1-3 [CONTAINS: GLUTAMINE-DEPENDENT CARBAMOYL-PHOSPHATE SYNTHASE (EC 6.3.5.5); ASPARTATE CARBAMOYLTRANSFERASE (EC 2.1.3.2); DIHYDROOROTASE (EC 3.5.2.3)]. | 6.00 | 241742.43 |
| 0.19 | 4 | P11978 | SIR4_YEAST | REGULATORY PROTEIN SIR4 (SILENT INFORMATION REGULATOR 4). | 9.03 | 152061.67 |
| 0.19 | 4 | P39057 | DYHC_ANTCR | DYNEIN BETA CHAIN, CILIARY. | 5.23 | 511782.89 |
| 0.19 | 4 | P20908 | CA15_HUMAN_1 | CHAIN 1: COLLAGEN ALPHA 1(V) CHAIN. | 4.91 | 153544.81 |
| 0.19 | 4 | P25391 | LMA1_HUMAN_1 | CHAIN 1: LAMININ ALPHA-1 CHAIN. | 5.92 | 335416.55 |
| 0.19 | 4 | P12003 | VINC_CHICK | VINCULIN. | 6.00 | 116867.67 |
| 0.19 | 4 | P98158 | LRP2_RAT_1 | CHAIN 1: LOW-DENSITY LIPOPROTEIN RECEPTOR-RELATED | 5.03 | 516748.40 |
| 0.19 | 4 | Q58445 | RPA1_METJA | DNA-DIRECTED RNA POLYMERASE SUBUNIT A' (EC 2.7.7.6). | 8.14 | 152780.97 |
| 0.19 | 4 | P10587 | MYSG_CHICK | MYOSIN HEAVY CHAIN, GIZZARD SMOOTH MUSCLE. | 5.46 | 228664.53 |
| 0.19 | 4 | P04146 | COPI_DROME | COPIA PROTEIN. | 8.73 | 162771.49 |
| 0.19 | 4 | Q66431 | RRPL_DUGBV | RNA-DIRECTED RNA POLYMERASE (EC 2.7.7.48) (L PROTEIN). | 6.90 | 459391.75 |
| 0.19 | 4 | P25823 | TUD_DROME | MATERNAL TUDOR PROTEIN. | 6.01 | 285238.86 |
| 0.19 | 4 | Q10411 | YD86_SCHPO | HYPOTHETICAL 222.8 KD PROTEIN C1F3.06C IN CHROMOSOME I. | 5.15 | 222786.78 |
| 0.19 | 4 | P36022 | DYHC_YEAST | DYNEIN HEAVY CHAIN, CYTOSOLIC (DYHC). | 5.90 | 471347.64 |
| 0.19 | 4 | P07751 | SPCN_CHICK | SPECTRIN ALPHA CHAIN, BRAIN (SPECTRIN, NON-ERYTHROID ALPHA CHAIN) (FODRIN ALPHA CHAIN) (SPTAN1). | 5.21 | 285363.39 |
| 0.19 | 4 | P12688 | YPK1_YEAST | SERINE/THREONINE-PROTEIN KINASE YPK1 (EC 2.7.1.-). | 6.07 | 76479.81 |
| 0.19 | 4 | P39812 | GLTB_BACSU | GLUTAMATE SYNTHASE [NADPH] LARGE CHAIN (EC 1.4.1.13) (NADPH-GOGAT). | 5.58 | 168857.24 |
| 0.19 | 4 | P13615 | RRPL_VSVJH | RNA POLYMERASE BETA SUBUNIT (EC 2.7.7.48) (LARGE STRUCTURAL PROTEIN) (L PROTEIN). | 8.85 | 241583.49 |
| 0.19 | 4 | Q57742 | HELX_METJA | PUTATIVE ATP-DEPENDENT HELICASE MJ0294. | 9.07 | 97267.97 |
| 0.19 | 4 | P48417 | CP74_LINUS_1 | CHAIN 1: ALLENE OXIDE SYNTHASE. | 5.86 | 53580.51 |
| 0.19 | 4 | P55824 | FAF_DROME | PROBABLE UBIQUITIN CARBOXYL-TERMINAL HYDROLASE FAF (EC 3.1.2.15) (UBIQUITIN THIOLESTERASE FAF) (UBIQUITIN-SPECIFIC PROCESSING PROTEASE FAF) (DEUBIQUITINATING ENZYME FAF) (FAT FACETS PROTEIN). | 5.74 | 307957.40 |
| 0.19 | 4 | Q09092 | SRK6_BRAOL_1 | CHAIN 1: PUTATIVE SERINE/THREONINE KINASE | 7.75 | 93484.45 |
| 0.19 | 4 | P27742 | ACVS_EMENI | DELTA-(L-ALPHA-AMINOADIPYL)-L-CYSTEINYL-D-VALINE SYNTHETASE (EC 6.-.-.-) (ACV SYNTHETASE) (ACVS). | 6.01 | 422456.49 |
| 0.19 | 4 | Q60429 | SRE2_CRIGR | STEROL REGULATORY ELEMENT BINDING PROTEIN-2 (SREBP-2) (STEROL REGULATORY ELEMENT-BINDING TRANSCRIPTION FACTOR 2). | 8.58 | 123655.22 |
| 0.19 | 4 | P30427 | PLEC_RAT | PLECTIN. | 5.71 | 533540.00 |
| 0.19 | 4 | P20028 | RPA2_DROME | DNA-DIRECTED RNA POLYMERASE I 135 KD POLYPEPTIDE (EC 2.7.7.6) (RNA POLYMERASE I SUBUNIT 2). | 8.63 | 128414.35 |
| 0.19 | 4 | P18296 | CUT1_SCHPO | CUT1 PROTEIN. | 7.58 | 209493.23 |
| 0.19 | 4 | P12954 | SRS2_YEAST | ATP-DEPENDENT DNA HELICASE SRS2 (EC 3.6.1.-). | 8.88 | 134325.24 |
| 0.19 | 4 | P08968 | RPC1_TRYBB | DNA-DIRECTED RNA POLYMERASE III LARGEST SUBUNIT (EC 2.7.7.6). | 9.01 | 170272.03 |
| 0.19 | 4 | P22168 | RRPO_FXMV | RNA REPLICATION PROTEIN (152 KD PROTEIN) (ORF 1) [CONTAINS: RNA- DIRECTED RNA POLYMERASE (EC 2.7.7.48); PROBABLE HELICASE]. | 6.77 | 152318.09 |
| 0.19 | 4 | P19787 | ACVS_PENCH | DELTA-(L-ALPHA-AMINOADIPYL)-L-CYSTEINYL-D-VALINE SYNTHETASE (EC 6.-.-.-) (ACV SYNTHETASE) (ACVS). | 5.57 | 421076.44 |
| 0.19 | 4 | P26046 | ACVT_PENCH | DELTA-(L-ALPHA-AMINOADIPYL)-L-CYSTEINYL-D-VALINE SYNTHETASE (EC 6.-.-.-) (ACV SYNTHETASE) (ACVS). | 5.69 | 425921.93 |
| Score: 0.43, 9 matching
peptides: P21694 (EFTU_SALTY)
pI: 5.30, Mw: 43152.36
ELONGATION FACTOR TU (EF-TU). |
|||||||
| user mass | matching mass |  mass (Dalton) mass (Dalton) |
#MC | modification | position | peptide | links |
|---|---|---|---|---|---|---|---|
| 1027.845 | 1027.5894 | -0.2555 | 0 | 270-279 | AGENVGVLLR | ||
| 1214.916 | 1214.6303 | -0.2856 | 0 | 304-313 | FESEVYILSK | ||
| 1233.917 | 1233.6163 | -0.3006 | 0 | 325-333 | GYRPQFYFR | ||
| 1390.988 | 1390.6485 | -0.3394 | 0 | METH: 56 | 45-56 | AFDQIDNAPEEK | |
| 1490.226 | 1489.8777 | -0.3482 | 0 | 124-136 | QVGVPYIIVFLNK | ||
| 1796.399 | 1795.9561 | -0.4428 | 0 | 8-24 | TKPHVNVGTIGHVDH GK | ||
| 1804.326 | 1803.8871 | -0.4388 | 0 | 59-74 | GITINTSHVEYDTPT R | ||
| 1962.52 | 1962.0218 | -0.4981 | 0 | 188-204 | IIELAGFLDSYIPEP ER | ||
| 1965.417 | 1964.96 | -0.4569 | 0 | 155-171 | ELLSQYDFPGDDTPI VR | ||
1 11 21 31 41 51 1 skekferTKP HVNVGTIGHV DHGKttltaa ittvlaktyg gaarAFDQID NAPEEKarGI 60 61 TINTSHVEYD TPTRhyahvd cpghadyvkn mitgaaqmdg ailvvaatdg pmpqtrehil 120 121 lgrQVGVPYI IVFLNKcdmv ddeellelve mevrELLSQY DFPGDDTPIV Rgsalkaleg 180 181 daeweakIIE LAGFLDSYIP EPERaidkpf llpiedvfsi sgrgtvvtgr vergiikvge 240 241 eveivgiket qkstctgvem frklldegrA GENVGVLLRg ikreeiergq vlakpgtikp 300 301 htkFESEVYI LSKdeggrht pffkGYRPQF YFRttdvtgt ielpegvemv mpgdnikmvv 360 361 tlihpiamdd glrfairegg rtvgagvvak vlgetc...
Un outils permettant une visualisation graphique des résultats, facile à utiliser et offrant une vaste gamme de fonctionalités complémentaires serait un gain de temps considérable et efficace. De plus, offrant une vision globale de chaque spectre de masses, il pourrait servir à des fins trés particulières de chaque chercheur ou département utilisant les mêmes banques de données.
Dans le cadre de ce projet, nous proposons une solution évolutive tout en utilisant des techniques classiques. En fait, BioGraph n'est qu'une de nombreuses applications possibles d'un noyeau constituant les 90% du code. En effet, il s'agit simplement de visualiser graphiquement des résultats de PeptIdent melangés à des données fournies par l'utilisateur et de les comparer entre elles. Le fonctionnement de BioGraph se résume ainsi:
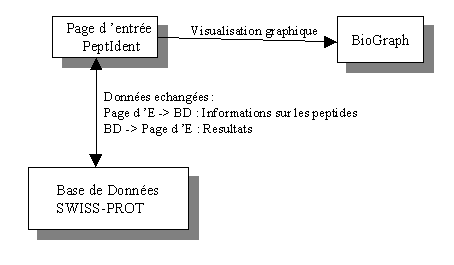
BioGraph génere deux spectres, le deuxième n'étant qu'un zoom entre deux valeurs specifiées de masses (PKM ou données utilisateur) du premier.Dans un fichier PKM, la première valeur (Center X) est la masse du premier peptide, la valeur Peak Y est son intensité relative dans la proteine digerée, Left X, Right X est le décalage (ou marge) min/max de la masse. BioGraph utilise les valeurs du fichier PKM, les affiche (bleu), puis, les mélange avec des valeurs generées par PeptIdent (les masses de peptides corréspondantes à celles dans la base de donnes de SWISS-PROT ainsi que d'autres informations telles que des missed cleavages ou des modifications post-transcriptionnelles) et les affiche avec une autre couleur (rouge/jaune). Les piques en jaune sont ceux des masses correspondantes avec une tolerance m-tol entre les deux séries de valeurs (respectivement, les piques en rouge sont ceux qui ne correspondent pas avec cette tolerance donnée).
Puisque PeptIdent ne genère pas d'intensités relatives, les valeurs d'un fichier PKM sont utilisées pour les masses correspondantes.
Voici un resultat produit par BioGraph :

On remarque une nette amélioration de visibilité par rapport aux résulats du PeptIdent. De plus, on peut effecuter des changements de tolerance (m-tol - par defaut, la valeurs utilisée par PeptIdent) On voit un premier avantage de BioGraph, il permet de réduire la tolerance de masses (m-tol) instantanement, ne montrant en jaune que les piques avec une masse dans cette tolerance donnée. Les piques rouges permettent ainsi de voir les resultats de PeptIdent avec une tolerance fixe et les piques jaunes avec une tolerance m-tol. De plus, puisqu'un spectromètre de masse peut faire des érreurs (d'envion 1 neutron) d'une manière uniforme (sur toutes les valeurs), x-ad permet de pondérer les masses avec une valeur unique.Le zoom (graphique du bas) permet de visualiser les deux résultats dans un champ de masses specifié (à droite). Les piques entourés en rouge sont simplement ceux qui correspondent à des piques jaunes sur le spectre complet.
Une importante fonctionalité de BioGraph est l'annotation de chaque peptide avec un maximum de valeurs connues. Si l'on pointe la souris sur un des piques, ces informations complémentaires (dans la mesure du possible, sa masse, son intensité, sa sequence en acides aminés, le nombre de missed-cleavages et les modifications post-transcriptionnelles) apparaissent dans un ToolTip.
Des options supplémentaires permettent d'afficher une grille du spectre (Grid), d'équilibrer les intensités sur les deux series de valeurs (Eq), d'effectuer un logarithme naturel sur toutes les intensités (Log, le logarithme conserve la propriété x>y -> log(x)>log(y) et vice-versa, ainsi accentuant et en equilibrant l'éspace occuppé par chaque pique sur le graphique), d'afficher les facteurs d'echelle x et y (Scale) et soit de spécifier l'échelle des masses (x) et des intensites (y), soit d'afficher les masses et les annotations des peptides correspondants. Un pourcentage de peptides correspondantes est également present.
Pour consulter l'aide on-line du BioGraph, clickez ici.
BioGraph à été ecrit en Java afin de permettre une utilisation relativement portable, à savoir en utilisant des OS variables et des navigateurs differents. Nous avons traité des aspects aussi variés que l'affichage de graphiques, l'adaptation automatique de l'échelle, le déplacement et le rafraichissement d'éléments independants ou encore le passage de paramètres entre l'utilisateur et notre programme.
Ainsi que décrit dans l'introduction, nous avons passé les 90% du temps a dévélopper un outil générique, puis d'utiliser ce travail afin de créer BioGraph. Ainsi, nous mettons à votre disposition une série de classes en Java et une methode fonctionnelle afin de permettre une réutilisation éfficace de cet ensemble pour des futurs projets semblables. Nous voyons un grand potentiel dans l'évolution de nombreux outils existants sur Expasy vers des solutions de plus en plus visuelles.
Nous tenons à remercier Elisabeth Gasteiger et Dr. Pierre-Alain Binz ainsi que Steven Gay pour un apport conséquent et une patience mise à rude epreuve quant à l'explication de certaines notions en bio-chimie.
Nous aimerions egalement citer l'utilisation d'algorithmes de Prof. Knuth pour le calcul d'echelle, tres gentillement mises a notre disposition par Pierre Sabatzus (Dataweb), ainsi que celle de la librairie MV4 (Vestris Inc.) pour le traitement CGI en C++.
| ExPASy Home page |
Site Map | Search ExPASy | Contact us | Proteomics tools |
| Hosted by NCSC US | Mirror sites: | Canada | China | Korea | Switzerland | Taiwan |