[Next]
[Previous] [Up] [Top]
4.0 Identification of Secondary Structure
4.1 Identification in 3D Structures
Three-dimensional protein structures at atomic resolution are now available
from both X-ray and NMR studies. Two important differences between structures
determined from these two methods are that 1.) protons are included (necessarily)
in the NMR solution structures and, with the exception of neutron diffraction
studies, they are practically invisible to X-ray techniques and 2.) X-ray
techniques provide a single, or at most a few, three-dimensional structures
present in the unit cell as a unique analytical solution to the experimental
data. On the other hand, structures determined by NMR methods are presented
as groups (10-50) of structures each satisfying the experimental constraints
equally well. This presents a potential problem in that either the entire
ensemble of structures are evaluated or a mean conformation is produced
and then evaluated. Mean structures from ill-defined portions of the polypeptide
chain will have non-standard geometries and may cause problems in analyses.
In any event, the lack of hydrogen in most crystallographically-determined
structures is not usually a problem as their positions are, in most cases,
uniquely defined by the polypeptide covalent geometry.
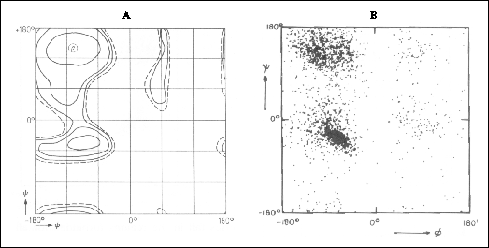
Angle plots
Right-handed alpha helices and beta sheets have very different backbone
dihedral angles (phi and psi) which appear in two separate regions of a
Ramachandran diagram (Figure 19 ). However, backbone dihedral
angles are seldom used for secondary structure identification. One reason
is that a given residue in a helical or extended conformation can have
backbone dihedral angles which differ considerably from the "typical" mean
values (and still be within the physically allowed regions). Another reason
for the lack of popularity of this method of secondary structure identification
is that extended conformations can exist (e.g. in loops) and not be part
of a sheet. We have already seen that the backbone dihedral angles near
the ends of helices are often irregular and the Ncap and Ccap residues
at helix boundaries contain non-helical phi, psi values and can make identification
difficult.

Figure 19. Ramachandran diagrams showing (A)
the potential energy distribution in the phi, psi plane for a pair of peptide
units with an Ala between and (B) a plot of the backbone dihedral angles
phi and psi of about 2500 residues in 13 proteins. Both (A) and (B) are
taken from Schulz & Schirmer, 1979. In (A), countours are drawn at
1 kcal/mole intervals from negative to zero (dotted).
Hydrogen bonds
It is most natural (and in practice most common) to identify regular
secondary elements (helix and sheet) based on the characteristic hydrogen-bonding
patterns (3.10 helix: i, i+3; alpha helix i, i+4 etc.). There are two obstacles
associated with identifying secondary structure from hydrogen bonds. One
is the criteria used for identifying a hydrogen bond itself, and the other
is the criteria used for identifying the secondary structure element (given
exact locations of all hydrogen bonds). Each deserves consideration.
There is no universally correct definition of a hydrogen
bond as there is no sharp border between the quantum-mechanical and
electrostatic regimes and no discontinuity in energy as a function of distance
or alignment that governs the interaction. From the analysis of small molecule
structures, an ideal hydrogen bond has a donor-acceptor distance of 2.9
Angstroms and a hydrogen-donor-acceptor angle of 0 degrees. Some criteria
commonly used in the literature are listed below. A hydrogen bond is identified
if:
-
the proton-acceptor distance is less than 2.4 Angstroms and the angle between
the proton-donor bond and the line connecting the donor and acceptor atoms
is less than 35 degrees (e.g., see Berndt et al., 1993).
-
the proton-acceptor distance is less than 2.5 Angstroms and the angle defined
by the hydrogen-acceptor-donor atoms lies between +/- 90 and 180 degrees
(Baker & Hubbard, 1984)
-
the energy defined by an electrostatic potential function is less than
a cutoff value (see Kabsch & Sander, 1983). These authors have proposed
perhaps the most generous criterium (E < -0.5 kcal/mol) allowing a distance
of up to 5.2 Angstroms between donor and acceptor at perfect alignment
and allowing a misalignment of up to 63 degrees at the ideal length (2.9
A).
Once the definition of a hydrogen bond is adopted and all such hydrogen
bonds in the protein under investigation are identified, the location and
extent of the secondary structural segments remain to be determined. In
principle, the core of alpha, 3.10, and pi helical segments should be unambiguous
due to the repeating (i, i+4), (i, i+3), and (i, i+5) hydrogen bonds, respectively.
However, as pointed out (3.1) the first
and the last helical turns each contain four residues in which only one
of the two potential backbone hydrogen bonds are formed. Are these first-turn
and last-turn residues also to be included in the helix? According to the
often used criteria of Kabsch & Sander (1983) in their "Dictionary
of Protein Structure" these residues are to be included in helical definitions
and set the minimum helical lengths to one turn.
Similarly, the central residues of beta strands are straightforward
to align into sheets whereas the end residues of each strand can contain
dihedral angles characteristic of extended conformations yet not participate
in the hydrogen bonding of the sheet (see Figure
9 ). To qualify as a strand of a beta sheet, most definitions require
the backbone amide nitrogen and carbonyl oxygen atoms of at least one residue
Despite all of the potential ambiguities listed above, regular
hydrogen bond patterns remain the most widely used and reliable method
of secondary structure identification
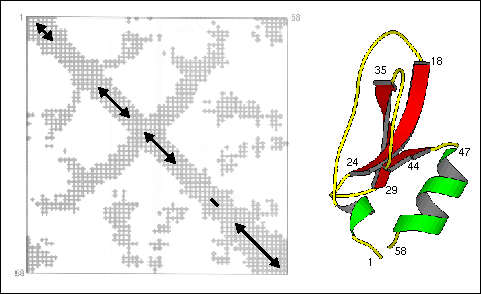
Distance plots
A two-dimensional plot of the distance between alpha carbons of residues
i and j is a useful way to present, in two-dimensions, the overall fold
of the polypeptide chain in the three-dimensional structure of a protein.
Such a plot is reproduced in Figure 20 where alpha carbons
closer than 10 Angstroms are indicated with a cross at the coordinates
corresponding to the residue numbers. In representations such as this,
helices can be identified as a strips directly adjacent to the diagonal,
antiparallel beta strands by strips perpendicular to the diagonal, and
parallel beta strands by off-diagonal strips parallel to the diagonal.
Contacts between secondary structures are also present in this representation.
The disulfide bond between residues 5 and 55 covalently attach the N- and
C-terminal segments producing the correlation between the two helical segments
3-6 and 47-58. However, while the general locations of helix and strand
segments can be obtained from distance plots, this method is no more reliable
than others for exact location of secondary structure boundaries.

Figure 20. Distance plot (contact map) of bovine
pancreatic trypsin inhibitor (left). Distances shorter than 10 Å
between alpha carbons are marked with a cross. The approximate positions
of the secondary structure elements are indicated on the diagonal (helix:3-6
and 47-58; sheet: 18-24, 29-35, and 45). Taken from Creighton, 1993. For
comparison, the three-dimensional structure of BPTI is shown with selected
residue positions labeled.
download 5PTI.PDB
download RasMol script
-
Angle plots
-
-
Hydrogen bonds
-
-
Distance plots
-
No Title - 31 MAY 96
written by Kurt D. Berndt
[Next] [Previous]
[Up] [Top]
Section 8 Index
Generated with CERN WebMaker