A platform for integrating threading results with other information
Florencio Pazos1, Burkhard Rost2 and Alfonso Valencia1 .
1 Protein Design Group. CNB-CSIC. Cantoblanco, Madrid 28049 Spain.
2 CUBIC Columbia University; Dep. Biochemistry & Molecular Biophysics; 630 West 168th Street; New York, N.Y. 10032; U.S.A.; rost@columbia.edu.
Running title: threadlize, a threading Interface.
Keywords: threading, TOPITS, graphic interface, protein structure prediction, correlated mutations.
Abstract.
Summary: We have developed a package for the interactive visualisation of results from different threading programs. Additionally, we have integrated relevant information about protein sequence, function, evolution, and structure into the interface.
Availability: A detailed documentation of THREADLIZE, and the binaries for IRIX, SunOS and Linux are available at http://www.cnb.uam.es/~pazos/threadlize. The package is free for academic users.
Contact: Alfonso Valencia (valencia@cnb.uam.es).
Supplementary information: http://www.cnb.uam.es/~pazos/threadlize
Automatic applications of threading (or fold recognition) methods result in fairly low levels of accuracy. However, in the last meeting for the critical assessment of structure prediction (CASP3), threading was shown to yield sustained levels of success (http://predictioncenter.llnl.gov/casp3/Casp3.html). The difference between the automatic application of threading and the actual results presented at the CASP3 meeting resulted from experts combining automatic threading results with results from other methods for sequence analysis. This success in combining outputs from different sources illustrates the need for interactive platforms simplifying such a task (e.g. Leplae et al., 1998; Miller et al., 1996). Here, we present our approach towards a general work-bench for the evaluation of threading results by assessment of structural and biological relevant information.
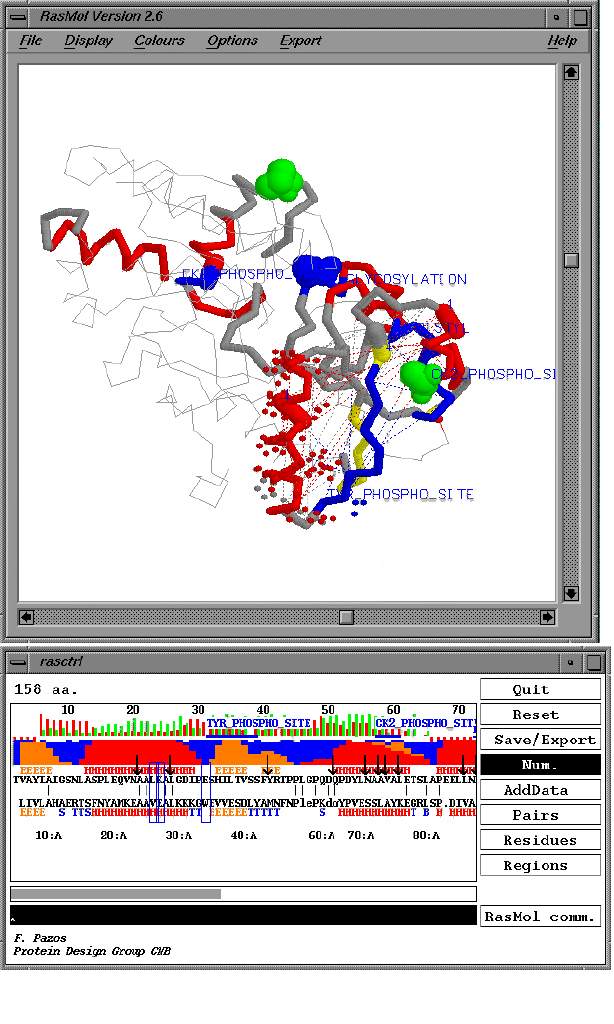
Threadlize uses as input information from two conceptually different threading programs, TOPITS (Rost, 1995; Rost et al., 1997) and Threader (Jones et al., 1992). and provides a general mechanism for integrating results from other programs. The program enables the simultaneous mapping of the following features onto the threading output: predictions of secondary structure (Rost et al., 1994) solvent accessibility (Rost et al., 1994), inter-residue contacts (in particular correlated mutations (Olmea & Valencia, 1997), physico-chemical residue scales (i.e. hydrophobicity, polarity, charge) and any other type of property provided by the user in a given format. Furthermore, the implicit threading model and related properties can be inspected visually through an interface with Rasmol v. 2.6. (Sayle & Milner-White, 1995) (Figure 1).
The Rasmol view of the protein model and the representation of the sequence-structure alignment are inter-connected. This enables operations like the selection of putative binding site residues in the sequence and their visualisation in the corresponding protein model. By default the regions covered by the threading alignment are highlighted in the model and in the sequence. This default view represents the initial protein model suggested by threading. In the alignment window (lower panel of Figure 1) the observed (for the protein of known structure, i.e. the template) and the predicted (for the query protein) secondary structure are compared. Optionally, the reliability of the secondary structure prediction and other residue-property scales can be optionally displayed, along with the regions matching Prosite patterns (Bairoch, 1992) or Prodom (Sonnhammer & Kahn, 1994) and coiled-coil patterns (Lupas et al., 1991). Additional structural information can be included by directly accessing SCOP (Murzin et al., 1995) or FSSP (Holm & Sander, 1994) databases of protein structure similarities. The analysis of this information is facilitated through an additional graphical representation (not shown).
The program uses the PDB database of protein structures (Bernstein et al., 1977), HSSP database of protein families (Sander & Schneider, 1993) and FSSP database of protein structure alignments (Holm & Sander, 1994) as retrieved from local copies of these databases (preferable) or directly them via Internet (slower), in plain or compressed format. Structural information about residues, pairs of residues or sequence regions can be imported from external files or can be generated inside the package. Two examples for such additional structural information are correlated mutations calculated with PLOTCORR (Pazos et al., 1997) and tree-determinant residues determined with SequenceSpace (Casari et al., 1995). The results of Threader are obtained from a local installation of the package (http://globin.bio.warwick.ac.uk/~jones/threader.html). The results of TOPITS are obtained from either a local installation of the program, or the public internet server PredictProtein (http://dodo.cpmc.columbia.edu/predictprotein). An interactive version accessing TOPITS results through WWW forms is in preparation.
Acknowledgements.
We happily acknowledge, Roger Sayle, for his help with Rasmol. Suggestions from the members of the “Protein Design Group” and the participants in the “Frontiers on Protein Structure Prediction” course are also acknowledged. We have extensively used the program during our participation in CASP (see; http://gredos.cnb.uam.es/pazos/working.html).
The interface allows the direct inspection of the list of threading models and the basic information provided in the PDB, SCOP or FSSP databases (not shown in the figure). From this sorted list users can proceed to analysing particular cases by opening the windows shown in the figure with the (button “Interactive Alignment”) option. The first window displays the sequence-to-structure alignment; the second window the displays the corresponding Rasmol view of the three-dimensional model implied by that alignment. The button “AddData” enables the representation of other data, like the secondary structure prediction reliability, different residue chemical properties or the predicted solvent accessibility.
The Rasmol command line is accessible by directly typing the commands or through the menu “Rasmol comm.” which items are configured by the user. The buttons “Residues”, “Pairs”, and “Regions” facilitate the import of data about individual residue properties, (e.g. sequence conservation values); residue-residue properties, (e.g. disulphide bridges or predicted contacts); or protein protein regions (e.g. Prosite patterns or Prodom domains). The button Save/Export” allows for various formats to save and print the display.
Bairoch, A. (1992). PROSITE: A dictionary of sites and patterns in proteins. Nucl. Acids Res. 20, 2013-2018.
Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. F., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., & Tasumi, M. (1977). The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535-542.
Casari, G., Sander, C., Valencia, A. (1995). A method to predict functional residues in proteins. Nature Structural Biology . 2, 171-178.
Holm, L., & Sander, C. (1994). The FSSP database of structurally aligned protein fold families. Nucl. Acids Res. 22, 3600-3609.
Jones, D., Taylor, W., & Thornton, J. (1992). A new approach to protein fold recognition. Nature . 358, 86-89.
Leplae, R., Hubbard, T. J. P., & Tramontano, A. (1998). GLASS: A tool to project protein structure prediction data into three-dimensions and evaluate their consistency. Proteins . 30, 339-351.
Lupas, A., Dyke, M. v., & Stock, J. (1991). Predicting coiled coils from protein sequences. Science . 252, 1162-1164.
Miller, R. T., Jones, D. T., & Thornton, J. M. (1996). Protein fold recognition by sequence threading: tools and assessment techniques. FASEB J . 10, 171-178.
Murzin, A. G., Brenner, S. E., Hubbard, T., & Chotia, C. (1995). SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol . 247, 536-540.
Olmea, O., & Valencia, A. (1997). Improving contact predictions by the combination of correlated mutations and other sources of sequence information. Folding & Design . 2, S25-S32.
Pazos, F., Olmea, O., & Valencia, A. (1997). A graphical interface for correlated mutations and other structure prediction methods. CABIOS . 13, 319-321.
Rost, B. (1995). TOPITS: Threading one dimensional predictions into three dimensional structures. In C. Rawlings & e. als (Ed.), Third International Conference on Intelligent Systems for Molecular Biology, (pp. 314-321). England, Menlo Park:
Rost, B., Sander, C., & Schneider, R. (1994). PHD - A mail server for protein secondary structure prediction. Comp. Applic. Biosci. 10, 53-60.
Rost, B., Schneider, R., & Sander, C. (1997). Protein fold recognition by prediction-based threading. J Mol Biol . 270, 471-480.
Sander, C., & Schneider, R. (1993). The HSSP data base of protein structure-sequence alignments. Nucl. Acids Res. 21, 3105-3109.
Sayle, R., & Milner-White, E. (1995). RASMOL: biomolecular graphics for all. Trends Biochem Sci . 20, 374.
Sonnhammer, E., & Kahn, D. (1994). Modular arrangement of proteins as inferred from analysis of homology. Protein Sci . 3, 482-492.