Patrones, perfiles
y dominios.
Parte
teórica
Por ejemplo, si dos proteínas se parecen en un 15 o un 20% de su secuencia (es decir, sólo coinciden en uno de cada 5 ó 6 aminoácidos) no sabremos si son homólogas, pero si resulta que este parecido se concentra en los residuos funcionales de la proteína (p.e. en el centro activo), la probabilidad de que tengan un origen evolutivo común es mayor. ¿Cómo podemos saber si las identidades se concentran en esos residuos importantes? Es lo que veremos hoy. La fuente principal son los alineamientos múltiples.
Estos aspectos son los que han motivado el surgimiento de métodos de comparación de secuencias (y de búsqueda de homólogos) más sofisticados, entre los que destacamos los patrones, los perfiles y los HMMs (hidden markov models o modelos de Markov ocultos).
En
esta clase explicaremos estos métodos y también los recursos
que podemos encontrar en la web para utilizarlos. Asimismo, veremos que
existen muchas bases de datos que utilizan estos métodos y ofrecen
mucha información. En la siguiente clase (Familias de proteínas)
hablaremos de las familias de proteínas, del interés que
tiene su clasificación y estudio y de cómo los perfiles y
otros métodos nos ayudan a identificarlas dentro del océano
de
secuencias que conocemos.
Secuencias consenso y expresiones regulares
Cuando consultamos la literatura referente a estos aspectos que vamos a tratar de las proteínas, encontramos que existe cierta laxitud y, a veces también mal uso, respecto a algunos términos como son "motivo" o "dominio". El segundo, el término dominio, lo introduciremos más adelante.
Los alineamientos múltiples son la fuente principal para determinar qué partes de la secuencia son más importantes para su función o estructura, y existen diversas aproximaciones para utilizar esta información.
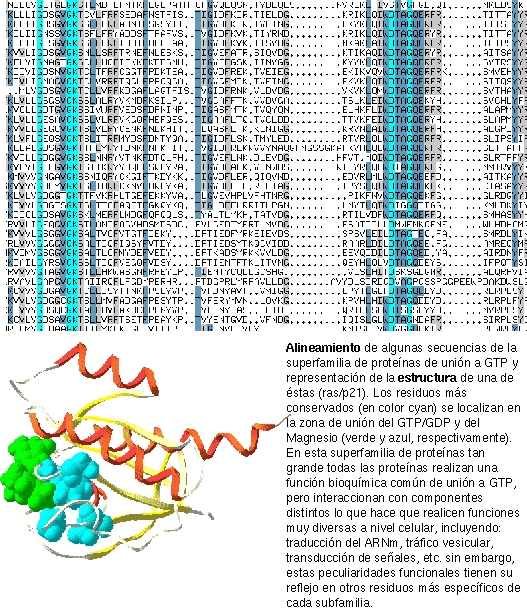
Motivo: si observamos un alineamiento múltiple de proteínas homólogas veremos que algunas columnas varían bastante, mientras que otras están más conservadas. Cuando observamos ciertas columnas cercanas con una alta conservación, es decir, cuando encontramos trocitos de las secuencias que se conservan más que otros y que podrían caracterizar funcionalmente a las proteínas, entonces solemos hablar de MOTIVOS. (ver ejemplo en el alineamiento de más arriba)
AGTVATVSCAGTSATHACIGRCARGSCIGEMARLACIGDYARWSC.........IGTVARVSC <= Ejemplo de secuencia consenso
Estas expresiones o patrones se pueden utilizar para caracterizar motivos, indicando qué posiciones son más importantes y cuáles pueden variar y qué variaciones pueden sufrir. Primero veremos cómo hablar en el lenguaje de las expresiones regulares.
[AC]-x-V-x(4)-{ED}
Este patrón significa: [Ala o Cys]-cualquiera-Val-cualquiera-cualquiera-cualquiera-cualquiera-{cualquier aa excepto Glu y Asp}< A-x-[ST](2)-x(0,1)-V
Este patrón debe encontrarse en posición N-terminal ('<') y significa: Ala-cualquiera-[Ser o Thr]-[Ser o Thr]-(un o ningún aminoácido de cualquier tipo)-Val<{C}*>
Este patrón lo cumplen todas aquellas proteínas que no contienen cisteínas. El * significa 'cero o más elementos'.
AGTVATVSC
AGTSATHAC
IGRCARGSC
IGEMARLAC
IGDYARWSC
.........
IGTVARVSC <= Ejemplo de secuencia consenso
[AI]-G-X-X-A-[RT]-[SA]-C
Primeramente definiremos el concepto de dominio, ya que tiene bastante relación con los perfiles, los cuales usualmente cubren una mayor parte de las secuencias que los motivos (se podría decir que los patrones son a los motivos lo que los perfiles a los dominios).
Dominio: el concepto de dominio define una unidad estructural independiente en las proteínas. Sin embargo se utiliza con cierta laxitud: por ejemplo, en estudios genéticos de deleción a veces se utiliza como sinónimo de la parte mínima de la secuencia capaz de realizar la función estudiada. En las bases de datos de dominios como PFam, un dominio se suele corresponder con el núcleo del dominio estructural, aquella zona más similar entre todas las proteínas de una familia, aunque no tiene por qué coincidir exactamente con los límites del dominio estructural.
A
continuación se muestra un alineamiento múltiple y cómo
se construiría el perfil según el método empleado
en PROSITE.
F K L L S H
C L L V
F K A F G Q
T M F Q
Y P I V G Q
E L L G
F P V V K E
A I L K
F K V L A A
V I A D
L E F I S E
C I I Q
F K L L G N
V L V C
A -18 -10 -1 -8 8 -3 3 -10 -2 -8
Como se puede apreciar, los pesos para cada aminoácido para cada columna no sólo se corresponden con sus frecuencias, sino también con sus propiedades físico-químicas, ya que se utiliza la información contenida en matrices como BLOSUM62 para completar la información. Por ejemplo, la A (alanina) de la tercera columna recibe una puntuación menor (-1) que la M (metionina) (+10), a pesar de que no haya ninguna metionina. Esto es porque la M es más parecida en sus propiedades a L, I, V y F que la alanina.
Los perfiles también son capaces de incluir información de inserciones y deleciones.
Alineamiento de una secuencia con un perfil:
S E Q U E N
C E
. . . . . .
. . .
p
. . . . . .
. . .
r \
. . _ . _ . . . .
. .
o
\
. . . . . .
. . .
f
\
. . . . . .
. . .
i
|
. . . . . .
. . .
l
\
. . . . . .
. . .
e
. . . . . .
. . .
Es similar al alineamiento de dos secuencias, pero no se comparan pares de aminoácidos o posiciones sino que cada aminoácido de la secuencia se compara con cada posición del perfil. Es decir, la puntuación no se obtiene de una matriz como BLOSUM, sino que viene implícita en el perfil con el que estamos alineando nuestra secuencia.
El
camino definido mediante las líneas horizontales, verticales y diagonales
describe el alineamiento:
S E Q U E - Nr - - o f i l
A
-18 -10 -1 -8 8 -3 3 -10
-2 -8
C
-22 -33 -18 -18 -22 -26 22 -24 -19 -7
D
-35 0 -32 -33 -7 6 -17 -34 -31
0
E
-27 15 -25 -26 -9 23 -9 -24 -23 -1
F
60 -30 12 14 -26 -29 -15 4 12 -29
G
-30 -20 -28 -32 28 -14 -23 -33 -27 -5
H
-13 -12 -25 -25 -16 14 -22 -22 -23 -10
I
3 -27 21 25 -29 -23 -8 33 19 -23
K
-26 25 -25 -27 -6 4 -15 -27 -26 0
L
14 -28 19 27 -27 -20 -9 33 26 -21
M
3 -15 10 14 -17 -10 -9 25 12 -11
N
-22 -6 -24 -27 1 8 -15 -24 -24 -4
P
-30 24 -26 -28 -14 -10 -22 -24 -26 -18
Q
-32 5 -25 -26 -9 24 -16 -17 -23 7
R
-18 9 -22 -22 -10 0 -18 -23 -22 -4
S
-22 -8 -16 -21 11 2 -1 -24 -19 -4
T
-10 -10 -6 -7 -5 -8 2 -10 -7
-11
V
0 -25 22 25 -19 -26 6 19 16 -16
W
9 -25 -18 -19 -25 -27 -34 -20 -17 -28
Y
34 -18 -1 1 -23 -12 -19 0 0 -18
supongamos que queremos alinear la secuencia FKTLGCCLLV:
El mejor alineamiento será:
F K L L S H
C L L V
F K A F G Q
T M F Q
Y P I V G Q
E L L G
F P V V K E
A I L K
F K V L A A
V I A D
L E F I S E
C I I Q
F K L L G N
V L V C
F K T L G
C C L L V
Y la puntuación: 60 25 -6 27
28 -26 22 33 26 -16
lo cual suma en total: 173.
Éste es un caso sencillo, sin incluir 'gaps', es decir, sin inserciones ni deleciones.
Cada puntuación tiene un significado estadístico, es decir, se pueden obtener p-values o e-values.
Los perfiles sin embargo son sensibles a determinados problemas. Por ejemplo, si existe un sesgo en la representación de las secuencias del alineamiento, es decir, si hay muchas secuencias demasiado parecidas y sólo unas pocas un poco más divergentes, entonces el perfil dará preferencia a encontrar secuencias del primer tipo. Existen formas de corregir estos sesgos, por ejemplo dando distintos pesos a las secuencias.
Por otra parte, en algunos casos es mejor usar patrones, especialmente cuando queremos describir motivos pequeños en los que no debería haber variaciones, por ejemplo, sabemos que en dicho motivo en la posición x debe haber un glutamato (E) y como tal lo expresamos en la expresión regular. Sin embargo, la flexibilidad del perfil podría tolerar que en esa posición un aspártico (D) obtuviera una puntuación positiva (E y D son muy parecidos en sus propiedades físico-químicas).
La limitación más importante
de los perfiles es que el sistema de puntuación que emplean tiene
una base estadística pobre.
Los modelos de Markov ocultos (HMMs: hidden Markov models) se desarrollaron inicialmente para el reconocimiento automático de voz. Este problema es similar al del reconocimiento de relaciones evolutivas entre las proteínas. Lo que se hacía era fragmentar el mensaje hablado en trocitos (frames) o sonidos aislados (de 10-20 milisegundos). Cada trocito o frame era automáticamente asignado a una de 256 categorías de sonido predefinidas (a la que más se pareciera). El resultado de esto era una cadena o larga secuencia de etiquetas de categorías a partir de las cuales realizar el reconocimiento de voz automático, detectando qué fonemas y qué palabras se han dicho. El problema es que existen grandes variaciones en la pronunciación, también en la duración de las distintas partes de la palabra.
Un HMM se entrena a partir de diversas observaciones en las que esperamos que las posibles variaciones se hayan producido, por ejemplo, para reconocer la palabra "vaca" u "otorrinolaringólogo" se debe entrentar el HMM a partir de muchas muestras de "vaca" y "otorrinolaringólogo", pronunciadas en distintos contextos o por distintas personas. El resultado es una estructura del siguiente modo:
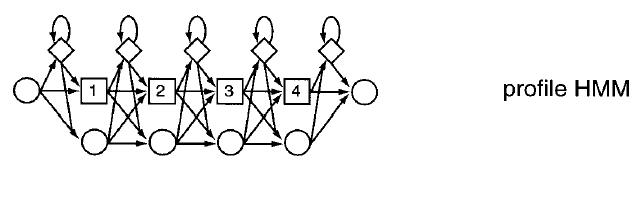
Un HMM está compuesto por una serie de nodos o estados cada uno de los cuales emite símbolos (una de las 256 categorías de sonido o uno de los 20 aminoácidos, por ejemplo) con una probabilidad dada (como en los perfiles). Los distintos estados están conectados secuencialmente existiendo probabilidades de transición entre ellos. Además existen probabilidades de inserción y deleción (en realidad éstas también se consideran estados). Eso es lo que son básicamente los HMMs, cuya principal ventaja es que tienen una base probabilística muy sólida.
Una vez entrenado un HMM para "vaca", cada vez que alguien diga algo y lo convirtamos en una cadena de etiquetas podremos determinar con qué probabilidad esa cadena podría ser emitida por el HMM de "vaca". En el caso de las secuencias lo tenemos más fácil incluso, ya que éstas ya vienen en forma de cadenas o secuencias.
Un ejemplo:
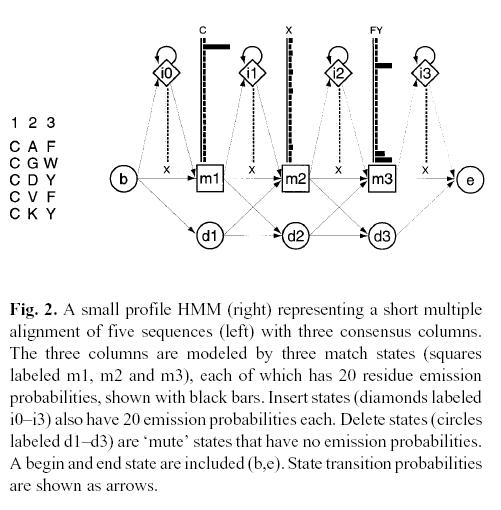
Por último señalar que éste es sólo un ejemplo del amplio abanico de aplicación de los HMMs, que pueden construirse con muy variadas arquitecturas y aplicarse para solucionar muy diversos problemas:

Búsquedas con secuencias intermedias (ISS: intermediate sequence searches)
Este método no utiliza información de qué posiciones son más importantes, ni utiliza alineamientos múltiples. Sin embargo, puede superar algunas de las limitaciones que tienen los métodos de búsquedas simples (BLAST, FASTA, ...).
Recordemos la limitación de BLAST: que no es capaz de distinguir entre parecidos que son reflejo de una relación de homología y parecidos que se producen al azar cuando el % de identidad está por debajo del ~25% (aprox.).
¿Cómo consiguen las búsquedas con secuencias intermedias
superar esta limitación? Gracias a que la homología entre
las proteínas presenta la propiedad transitiva, esto es:
si la proteína A es homóloga a la proteína B, y B
es homóloga a C, entonces A también es homóloga a
C (aunque A y C no se parezcan). Esto sólo es cierto cuando las
zonas homólogas se corresponden, es decir, sólo se aplica
a nivel de dominios.
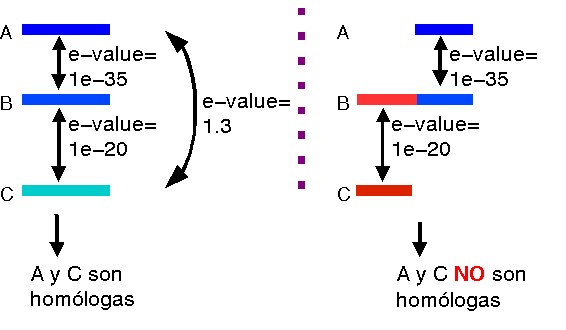
Por tanto, si realizamos búsquedas recursivas con los homólogos que vamos encontrando quizás seamos capaces de encontrar homólogos remotos, pero su éxito depende de que existan secuencias intermedias a distancias suficientemente cercanas (por ejemplo con % de identidad de secuencia > 30%). El método es como ir saltando de secuencia en secuencia. Más adelante veremos que, además de la detección de homólogos remotos, este método tiene otras utilidades para clasificar las proteínas en familias.
Ejemplo:

Algunas bases de datos de patrones, perfiles y hmms. Prosite y Pfam.
A continuación
describiremos las bases de datos PROSITE y PFam, de forma que podamos realizar
la práctica con ellas. En la siguiente lección hablaremos
de las familias de proteínas y en ese contexto volveremos a hablar
de esas bases de datos y de otras similares.
La versión 18.20 de Prosite, del 2 de febrero de 2004, contiene 1.245 entradas de documentación, las cuales describen 1691 patrones, reglas (rules) y perfiles. En http://us.expasy.org/prosite/prosuser.html hay una descripción de la base de datos.
Y en http://us.expasy.org/cgi-bin/nicedoc.pl?PDOC50020 hay un ejemplo de una entrada de documentación.
La construcción de los patrones es manual: a partir de revisiones bibliográficas acerca de familias de proteínas, se consultan los alineamientos múltiples y se derivan expresiones regulares. La eficiencia (sensibilidad/especificidad) de estos patrones es comprobada aplicándolos a las secuencias de la base de datos Swiss-Prot (la cual está anotada por expertos). Si el patrón no es satisfactorio se intenta refinar. En este procedimiento se intenta generar patrones los más cortos posibles.
En Prosite también existen perfiles. Se construyen a partir de los mismos alineamientos múltiples que los patrones, para intentar superar las limitaciones de las expresiones regulares.
Existen dos tipos de entradas en Prosite: las que definen los patrones y los perfiles y las que contienen la documentación.
La estructura de una entrada de Prosite es ésta:
ID Identification (Begins each entry; 1 per entry)
'A': archaea'B': bacteriofagos'E': eucariotas'P': procariotas o bacterias'V': virus de eucariotas
(hay más información del formato de PROSITE aquí).
Tour: Prosite:
(browse,
search,
scanprosite,
motifscan,
pratt)
Pfam (http://www.sanger.ac.uk/Software/Pfam/index.shtml) es una base de datos de perfiles tipo HMM. Se divide en dos partes: pfam-A y pfam-B. La primera se construye manualmente: cada vez que se identifica una nueva familia de proteínas un experto elabora un HMM diagnóstico: un HMM capaz de detectar a todas las otras proteínas de la familia y sólo a éstas. Por otra parte, como pfam-A sólo cubre el 73% de Swiss-Prot y TrEMBL, existe pfam-B. Ésta se genera automáticamente a partir de aquellos perfiles que existen en PRODOM (también generados automáticamente) que no se corresponden con ningún pfam-A. Un 21% de las proteínas de Swiss-Prot y TrEMBL presentan al menos un pfam-B.

Pfam-A contiene 7.316 familias de proteínas (bastante más que Prosite).
Pfam trabaja con dominios, es decir, cada perfil HMM se corresponde con un dominio, aunque no necesariamente cumplan la definición de dominio estructural independiente, sino más bien suelen ser regiones características de una determinada familia de proteínas.
Además de las ventajas que de por sí tiene esta clasificación, pfam resulta útil para:
-analizar los alineamientos múltiples que contiene.Tour: Pfam: (browse, protein search, dna search, taxonomy). En el ejemplo de CARD (http://www.sanger.ac.uk/cgi-bin/Pfam/getacc?PF00619) podemos ver algunas de las características de una entrada de Pfam.
-estudiar la organización de dominios de las proteínas.
-examinar la distribución filogenética de las proteínas que presentan el dominio.
-también permite ver la estructura tridimensional de los dominios, cuando ésta se conoce.
-y, de las más importantes, permite buscar con una secuencia de una proteína empleando los métodos de HMM, que hoy por hoy son los más eficaces en el análisis de secuencias.
Búsqueda
en bases de datos. PSI-BLAST y HMMer
A los programas de BLAST, PSI-BLAST, PHI-BLAST.... se puede acceder desde el sitio del NCBI.

Además de este modo de funcionamiento básico a partir de una secuencia, PSI-BLAST también puede iniciarse con un alineamiento múltiple.
Ejecutarlo localmente, por línea de comandos, puede resultar un poco tedioso, y resulta más sencillo utilizar el servidor del NCBI, que además ofrece algunas ventajas: por ejemplo, permite seleccionar cuáles de los homólogos deberán ser utilizados para construir el perfil.
Ejemplo (el que se sigue en el tutorial
del NCBI)
>gi|2501594|sp|Q57997|Y577_METJA PROTEIN MJ0577
MSVMYKKILYPTDFSETAEIALKHVKAFKTLKAEEVILLHVIDEREIKKRDIFSLLLGVAGLNKSVEEFE
NELKNKLTEEAKNKMENIKKELEDVGFKVKDIIVVGIPHEEIVKIAEDEGVDIIIMGSHGKTNLKEILLG
SVTENVIKKSNKPVLVVKRKNS
PHI-BLAST (Pattern-Hit Initiated BLAST): permite realizar búsquedas que son mezcla de búsquedas de patrones y alineamiento de secuencias. Se inicia con una secuencia y un patrón (que presenta dicha secuencia y que es característico de dicha familia). Lo que hace es buscar todas aquellas secuencias que presenten ese patrón, pero para descartar aquellas ocurrencias del patrón que se producen al azar (especialmente con patrones cortos), además comprueba que las regiones vecinas al patrón sean parecidas entre las secuencias.
Instalación local
hmmbuild => hmmcalibrate => hmmsearch
