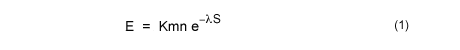| volver al índice |
Alineamiento de secuencias. Búsqueda
de parecidos. Alineamientos múltiples
Parte teórica
Federico Abascal
La mayoría de las imágenes (e ideas)
están tomadas de las clases
de Paulino Gómez Puertas (enlace),
Oswaldo Trelles (enlace)
y Ramón Alonso Allende (enlace).
¿Qué es
una secuencia?
es una serie de elementos encadenados uno detrás de otros, por
eso hablamos de secuencias de nucleótidos y de secuencias de aminoácidos.
A través de letras podemos identificar los distintos monómeros
que conforman la macromolécula (ejemplo: A: adenina; T: timina;
C: citosina; y G: guanina; ó A: alanina; D: aspártico; E:
glutámico; etcétera).
¿Por qué
comparar secuencias?
Para comprender la utilidad de la comparación de secuencias
y de la búsqueda de homólogos (proteínas que
tienen un origen evolutivo común), necesitamos tener algunos conceptos
acerca de cómo evolucionan las proteínas.
Breve introducción acerca de cómo evolucionan las proteínas.
La idea generalmente aceptada es que a lo largo de la evolución unas especies van dando lugar a otras nuevas. Detrás de estas especiaciones está la variación genética de los organismos, con la evolución de los genomas, sus genes, y de las proteínas codificadas por éstos.
Básicamente podemos distinguir las siguientes formas en que los genes (y por consiguiente las proteínas) evolucionan:
*mutación: cuando el ADN se replica se pueden producir errores al realizar la copia; también pueden introducirse mutaciones por efecto de agentes externos (mutagénicos) como por ejemplo la luz ultravioleta o ciertos agentes químicos.
*duplicación: cuando un gen se duplica se abre una puerta para la adquisición de nuevas funciones biológicas: las mutaciones en el nuevo gen son más fácilmente tolerables. Las más de las veces estos genes terminaran degenerando, convirtiéndose en pseudogenes.
*barajado de dominios: muchas proteínas están constituidas por entidades estructurales independientes, a las que llamamos dominios. Mediante recombinación se puede producir un barajado de dominios. Por ejemplo, a partir de la combinación de dominios ya existentes se pueden obtener nuevas proteínas, cuya función será el resultado de las sub-funciones de sus dominios. Es como jugar al lego.
Cuando observamos diferencias entre secuencias de proteínas que sabemos que tienen un origen evolutivo común, estas diferencias están hablándonos de las propiedades funcionales de las proteínas: algunas diferencias estarán allí porque la mutación no ha alterado la función, han sido más o menos neutrales; la observación de otras diferencias quizás tenga que ver con el hecho de que esas proteínas, aunque tengan un origen común, realicen funciones distintas. Y otras diferencias tendrán que ver con el modo de vida del organismo: por ejemplo, las bacterias que viven en fuentes termales a temperaturas muy altas tienen proteínas con una T (temperatura) de desnaturalización también muy alta (gracias a la secuencia de aminoácidos). Por otra parte, el hecho de que determinadas posiciones de las secuencias permanezcan invariables, nos indica que éstas tienen una especial importancia para el mantenimiento de la estructura o la función de la proteína.¿Cómo responde un organismo a una mutación? (dicho de otra forma: ¿cómo le afecta una mutación?): de distinta forma según las distintas situaciones. Por ejemplo, en cuanto a las mutaciones, éstas se tolerarán cuando no produzcan alteraciones en la estructura o en la función de la proteína. Sin embargo, en el caso de que se trate de un gen duplicado existe una mayor flexibilidad, la pérdida de función del nuevo gen no es dramática ya que existe otra copia. El barajado de dominios en unos casos producirá proteínas beneficiosas y en otros no. Según lo beneficiosos o perjudiciales que sean los cambios en las proteínas los organismos que los porten serán seleccionados positiva o negativamente, lo que determinará que los cambios pasen a generaciones venideras o, visto de otra forma, determinará que en el momento actual, en el que vivimos nosotros, observemos esos cambios: las secuencias que observamos ahora reflejan toda una historia evolutiva en la que las proteínas han ido divergiendo, adquiriendo nuevas funciones, adaptándose a nuevos entornos... y tenemos la oportunidad de desvelar algunos de esos secretos.
Las proteínas son cadenas polipeptídicas de aminoácidos ensamblados secuencialmente (de ahí el nombre de secuencia). La secuencia de una proteína determina qué estructura tridimensional tendrá ésta. El resultado de la adopción de una estructura tridimensional es que se forman superficies moleculares con distintas propiedades, quedando aminoácidos específicos en una orientación determinada, lo que permite a la proteína llevar a cabo su función.
Entonces, podemos
decir que la comparación de secuencias es una forma de hacer
arqueología, de descubrir qué partes de las secuencias son
más importantes (están más conservadas), descubrir
qué proteínas tienen un origen común (existen modelos
estadísticos que nos ayudan a distinguir parecidos al azar de parecidos
que reflejan un mismo origen evolutivo), la comparación de secuencias
también nos puede servir para predecir la estructura de las proteínas
(las proteínas homólogas tienen una misma arquitectura tridimensional),
o también nos puede ayudar a predecir la función de las proteínas
(aunque en este aspecto hay que ser cautelosos ya que a lo largo de la
evolución proteínas con un origen común pueden terminar
desarrollando distintas funciones, como veremos más adelante).
|
TCAGACGATTG (r=0)
ATCGGAGCTG
TCAGACGATTG (r=1)
ATCGGAGCTG
TCAGACGATTG (r=0)
ATCGGAGCTG
TCAGACGATTG (r=2)
ATCGGAGCTG
...
TCAGACGATTG
(r=4) éste sería
el mejor alineamiento
ATCGGAGCTG
...
TCAGACGATTG (r=0)
ATCGGAGCTG
|

Matriz para encontrar el mejor alineamiento |

Matriz dot-plot. Las diagonales representan zonas que alinean bien. |
Caso 2:
ATGAGATG----ATGATAACGATG
ATGATGATGTATAGATAACGAATG
Penalizando de igual modo apertura y extensión de gaps (huecos)
en ambos casos la penalización tendría el mismo valor.
Si penalizamos más fuertemente la apertura que la extensión
de un gap, el caso 2 será más probable evolutivamente (tendrá
una puntuación más favorable, menos penalización).
Para elegir en cada casilla de la matriz cuál es la mejor opción,
se elije el máximo de:

Esta forma de "rellenar" la matriz garantiza que obtenemos el mejor
alineamiento, sin necesidad de explorar todas las posibilidades.
Un ejemplo: las secuencias:
HEAGAWGHEE
y
PAWHEAE



Búsquedas de parecidos en las bases de datos.
Actualmente las bases de datos contienen un gran número de secuencias, y crecen de forma exponencial. Por ejemplo en Genbank ya hay: más de 28.000 millones de pares de bases (nucleótidos), correspondientes a más de 22.000 millones de secuencias. Por otra parte conocemos más de un millón de secuencias de proteínas.
En este contexto, aplicar algoritmos como el de Smith & Waterman
no es factible, ya que tardaría demasiado tiempo, a no ser que dispusiéramos
de máquinas especiales que trabajasen en paralelo. Por eso existen
métodos como BLAST y FASTA que aplican heurísticas
(o "truquillos") para reducir el tiempo de búsqueda. Estos "truquillos"
no garantizan el resultado óptimo pero casi siempre funcionan, y
la ganancia en tiempo hace que compense usarlos.
Heurísticas de BLAST y FASTA.
Estos métodos son muy rápidos. Básicamente utilizan los siguientes "truquillos":
- tablas de dispersión: en lugar de representar una secuencia como tal, utilizan una tabla tal que:
posición : 12345678901
secuencia X: TCAGACGATTGTabla de disperisón de X:
A 3, 5, 8
C 2, 6
G 4, 7, 11
T 1, 9, 10
Si hacemos lo mismo con una secuencia Y, ahora cuando comparemos X e Y sólo tendremos que recorrer una vez la secuenca Y, y por cada elemento de ella, tantas posiciones como se indique en la tabla de dispersión de X. Dicho de otro modo, si Y empieza por una C, no tenemos que mirar una a una todas las posiciones de X para ver donde hay una C, ya que ya hemos construido una tabla y podemos ver directamente, en la fila de las C, que en X hay una C en las posiciones 2 y 6.
- k-tuplas: en lugar de representar una secuencia de proteínas a partir de los 20 aminoácidos posibles, la representan mediante palabras de mayor tamaño, por ejemplo k=2, con lo que el alfabeto aumenta a 400 (20*20). De este modo, la tabla de dispersión tiene más filas (400), pero el número de columnas por fila disminuye, y por tanto el número de coincidencias entre X e Y también disminuye.
Estimación de la probabilidad de que un determinado parecido entre secuencias se deba al azar: el e-value
- Búsqueda en las diagonales en las que más probablemente se encuentra el mejor alineamiento: la idea es no consultar toda la matriz para buscar el mejor alineamiento sino fijarnos sólo en las diagonales que, por ejemplo, haya más coincidencias.
La mejor diagonal (la -1) (en este ejemplo no se tiene en cuenta la influencia de los gaps) es la correspondiente a:
GTCCGACTAGTG
|| || |
CATCGGAGCTG
La búsqueda del mejor alineamiento la haríamos quizás en las diagonales -1, 2 y 4, olvidándonos del resto. No garantiza el resultado óptimo pero casi siempre funciona, y es mucho más rápido.Gracias a estos truquillos, el tiempo de comparación de O(NxM) se reduce a O(N+M).
En FASTA (Lipman & Pearson, 1985) se obtienen las mejores diagonales y a partir de ellas se calculan los mejores alineamientos. Posteriormente se intentan unir las diagonales entre sí incluyendo gaps.
El artículo original de BLAST (Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, 1990) es el más citado de la década de los 90. La principal diferencia con FASTA es el modo en que busca las diagonales. En el caso de FASTA, si usamos una tabla de dispersión con k-tuplas de tamaño 4, sólo se tienen en cuenta las coincidencias entre las secuencias X e Y (p.e, la palabra AAAA en X sólo se corresponderá con AAAA en Y, mientras que si en Y hay AAAT, no se registrará el parecido). Sin embargo en BLAST, cuando se construye la tabla de dispersión de la proteína problema se pre-calculan los parecidos entre las distintas palabras. Por ejemplo: si la proteína problema (X) empieza por CTDCGRSGLI...) y usamos tuplas de k=4, la primera palabra de X será CTDC, y BLAST calculará qué otras palabras (según la matriz de sustitución) tienen una puntuación positiva (por ejemplo CSDC). De este modo se gana en sensibilidad, aunque aumenta el espacio de búsqueda. Para reducir un poco este espacio, sólo se apuntan aquellos parecidos entre palabras suficientemente altos (esto viene dado por un parámetro T de BLAST).
Uno de los problemas más importantes una vez hemos encontrado parecidos en las bases de datos es saber si son significativos o si podrían deberse al azar y por tanto no reflejan una relación de homología. En el caso de parecidos muy claros, la respuesta es evidente, pero hay muchos otros parecidos que no lo es, y para resolver este problema se han desarrollado diversos modelos estadísticos. Básicamente lo que se hace es calcular la probabilidad de que un alineamiento entre secuencias no relacionadas (por azar) alcance una puntuación (score) determinado; en esto influyen diversos aspectos:
- la matriz de sustitución empleada: hay matrices que tienden a dar puntuaciones más positivas que otras.
- la composición de aminoácidos de las secuencias alineadas: por ejemplo, si ambas secuencias tienen muchas Cys y éstas en la matriz de sustitución suelen tener puntuaciones más positivas que otros aminoácidos, una puntuación alta de tal alineamiento será menos significativa.
- la longitud de las secuencias alineadas (o el tamaño de la base de datos -ver más abajo-): cuanto mayores sean las secuencias, mayor será la probabilidad de que por azar alcancen un determinado score.
A partir de un modelo en el que estudiaron qué scores alcanzaban los alineamientos de secuencias generadas al azar (según las frecuencias observadas de aminoácidos), Karlin & Altschul desarrollaron la siguiente fórmula para el cálculo del e-value:
El e-value (E) de un determinado score indica cuántos alineamientos esperamos que por azar alcancen un score igual o mayor dadas los tres factores antes mencionados (no confundir con el p-value, que indica la probabilidad de que un score se haya alcanzado por azar al menos en una ocasión; e-value y p-value se relacionan mediante la siguiente fórmula:
y tienen un valor prácticamente idéntico en la escala que va de 0 a 0.01).
Cuando tratamos con una base de datos más importante que la longitud de las secuencias es el tamaño de la base de datos: cuanto mayor sea ésta, con mayor probabilidad aparecerán alineamientos que por azar alcancen un determinado score. En la fórmula del e-value K y lambda son dos parámetros que se determinan empíricamente, M y N son las longitudes de las secuencias y S es el score (la puntuación del alineamiento). Pues bien, en el caso del cálculo de e-values en el contexto de la búsqueda en bases de datos M se toma como el tamaño de la base de datos (número total de aminoácidos o nucleótidos; si bien se aplica una corrección -edge effect o efecto de los extremos- que se explica en el enlace: http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html).
En la práctica tenemos que tener en cuenta lo siguiente: el e-value depende del tamaño de la base de datos (si una DB es 10 veces menor que otra, el e-value será 10 veces menor, también). En general con bases de datos grandes, nos podemos fiar de e-values menores de 1e-05, y en el rango 1e-05 a 0.1 casi siempre nos podemos fiar; por encima de 0.1 ya es más arriesgado. Sin embargo, lo mejor siempre es utilizar el criterio propio, mirar los alineamientos a ojo, hacer alineamientos múltiples, búsquedas con PSI-BLAST, etcétera, como veremos más adelante.
Para saber más acerca de Alignment Scoring Statistics:Filtros SEG, XNU, COILS y DUST.http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html
http://blast.wustl.edu/doc/infotheory.htmlHay algunos casos en que las secuencias, por tener características especiales, pueden alcanzar fácilmente puntuaciones altas en sus alineamientos aún no estando relacionadas:
- secuencias de baja complejidad (filtro SEG para aminoácidos y filtro DUST para nucleótidos): por ejemplo secuencias con muchas alaninas o prolinas repetidas. Éstas secuencias darán fácilmente alineamientos de alta puntuación con otras secuencias que también tengan por ejemplo zonas ricas en prolinas, aunque no estén relacionadas. Además, casi cualquier forma de alinearlas dará puntuaciones elevadas. Por ejemplo:
secuencia 1: RCTAASAAAAAARAA
secuencia 2: GYAAAAALAAAAAAAlineamientos con buenas puntuaciones:
RCTAASAAAAAARAA
GYAAAAALAAAAAA
RCTAASAAAAAARAA
GYAAAAALAAAAAA
¿Cuándo aplicar los filtrados? Normalmente lo mejor es probar a realizar la búsqueda (p.e. BLAST) con y sin filtrado, inspeccionar los resultados y determinar si el filtrado en ese caso nos permite eliminar parecidos espúreos (al azar). Si no aporta nada, mejor no utilizarlo porque estamos perdiendo información de secuencia que podría ayudarnos a encontrar homólogos más lejanos.
RCTAASAAAAAARAA
GYAAAAALAAAAAA etcétera
El filtro SEG enmascara estas regiones sustituyendo los códigos de los aminoácidos por X.
- secuencias repetitivas (filtro XNU): se aplica a secuencias con cortas repeticiones.
- secuencias de coiled-coils (filtro COILS): se aplica a secuencias de coiled-coils. Éstas, al tener una periodicidad (suelen tener una Leu o una Ile cada 7 residuos) pueden dar buenas puntuaciones con otras proteínas que también adopten coiled-coils, sin que por ello compartan un origen evolutivo común (sean homólogas).
Por alineamiento múltiple nos referimos al alineamiento de más
de dos secuencias. Los alineamientos múltiples de secuencias son
el pilar central de multitud de métodos bioinformáticos.
Como hemos visto, el alineamiento mediante programación dinámica
de dos secuencias tiene una complejidad O(NxM). Pues bien, si queremos
alinear tres secuencias, la complejidad sería de O(NxMxL). O dicho
de otra forma: si alinear dos secuencias de 300 residuos tarda un segundo,
alinear tres secuencias tardará 300 segundos. Y alinear 10 secuencias
tardará 3008 segundos (más que la edad del universo).

Matriz tridimensional para el alineamiento de tres secuencias
(para cuatro secuencias necesitaríamos una matriz
de cuatro dimensiones)
En resumidas cuentas, no podemos resolver el problema del mismo modo que para un par de secuencias.
Una aproximación a la solución de este problema la propusieron Carrillo & Lipman en 1988, y posteriormente fue aplicada para el desarrollo del programa MSA (Lipman, Altschul & Kececioglu, 1989). La idea es calcular el parecido entre todos los pares de secuencias, de modo que obtengamos una medida de su distancia evolutiva. Se toman las dos secuencias más cercanas y se alinean entre sí. A partir de este momento ambas secuencias se tratan como una sola. Se vuelven a alinear las dos más cercanas.... y así sucesivamente hasta que no quedan más.
ClustalW (Thompson, Higgins & Gibson, 1994) es el programa más comúnmente utilizado y sigue un enfoque similar: a partir de la matriz de distancias entre las secuencias se construye una especie de árbol guía y se van alineando las secuencias entre sí de acuerdo a este árbol.
El resultado de estos programas no tiene por qué ser el óptimo, pero normalmente funcionan bien, aunque a veces hay que corregir algunas zonas del alineamiento a mano, utilizando programas como SeaView.
Finalmente, decir que recientemente se ha desarrollado un nuevo método
de alineamiento llamado T-coffee (Notredame,
Higgins & Heringa, 2000) que resuelve muchos de los problemas de ClustalW,
aunque requiere un mayor tiempo de computación.
¿Cuál es la utilidad de los alineamientos múltiples? lo veremos en la siguiente clase: Análisis de secuencias: motivos y perfiles.
| volver al índice |