Introduction to
Molecular Graphics on Desktop Computers
© 1997, 1998, 1999 Jean-Yves
Sgro UW-Madison
Contents
Introduction
-
Where to find
coordinates: The Protein Data Bank (PDB) database
-
The PDB database
-
Reference
-
Availability
-
PDB file names
-
Retrieving PDB files
-
The PDB file format
-
Example of 2 ATOM records
-
List of records.
-
Example of a PDB file
-
Coordinates and reference axes
-
Other formats
-
Desktop computer
graphics programs
-
Introduction
-
Graphical representations
-
RASMOL
-
Main features
-
Graphics window
-
Menu bar
-
Command line
-
Summary tables
Introduction
The visualization techniques of the structure
of macromolecules are companion tools to the sequence analysis algorithms.
New DNA sequences are being cloned and sequenced rapidely but the structure
of the putative encoded proteins cannot be determined based only on their
sequence. As the number of protein structures solved by x-ray crystallography
is increasing it will become easier to find structural homologues to fit
onto newly protein sequences. Molecular graphics play a key role in understanding
current structures and creating (structural) models.
Molecular graphics have evolved over the
last 30 years from a simple vector display on a high performance oscilloscope
to sensor based virtual reality. Some desktop computers are now more powerful
than mainframes of the last decade and there are free and commercial software
programs to manipulate 3 dimensional structures for the creation of publication-
quality images to illustrate research papers, proposals and to help visualize
target molecules, their structural properties or their interaction with
other molecules or ligands.
To be able to manipulate 3 dimensional
structures on a desktop computer with a molecular graphics program is critical
for today's molecular biologist and a necessary complement to sequence
analysis projects.
| Goal: During this session you will familarize
yourself with the program
RasMol. Your knowledge of RasMol will
allow you to manipulate and explore in detail existing 3 dimensional structures.
The outcome of this exploration will be a better understanding of structures
and can help you with the creation of a figures or animations. |
This material supplements the exercises and the program manuals.
But where do 3 dimensional structures come from ?
Biochemists and crystallographers have developped techniques to crystalize
macromolecules. Indeed proteins, nucleic acids or their complex can form
crystals in specific biochemical conditions. The crystals are very fragile
and small (often less than a millimeter) but they still can be placed inside
an x-ray beam. Because of the regular arrangement of the molecules within
the crystals the x-ray will diffract in a very specific pattern which can
be recorded on x-ray photographic film. With the help of powerful computer
and programs, the mathematical analysis of the diffraction pattern allows
the crystallographer to calculate where the electrons (of the atoms) of
the protein should be located in 3D space inside the crystal. They then
fit a wireframe representation of the amino acids inside the eclectron
density. When the position of the atoms is refined, the structure is published
and usually deposited at the Protein Data Bank at Brookhaven. These are
the structures that you can fetch with Netscape and display inside on your
desktop computer. A notable exception is for structures determined in the
private sector, these coordinates are proprietary are the authors are not
obligated to submit their data.
There are a lot of months or years of
work for each solved structure!
 |
| L(+) lactate dehydrogenase
crystals. Bar=100 µm. Ostendorp et al.(1996) Protein Science
5, 862 |
|
 |
| pentalenene synthase crystals. Lesburg et al. (1995)
Protein
Science 4, 2436 |
|
 |
| phosphoribulokinase crystals. Roberts et al. (1995)
Protein
Science 4, 2442 |
|
| Crystals are placed into an x-ray beam. The atoms of
the proteins within the crystals diffract the incident x-ray and create
diffraction
patterns on a film. With complex mathematical calculations crystallographers
obtain an electron density map into which the amino acid sequence
is fitted with help of computer graphics. |
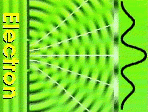 |
| Diffraction Amplitude waves of diffractied electrons
can add or substract to each other. The result are the white dots on the
diffraction image. |
|
 |
| Diffraction image from a pentalenene synthase crystal.
Lesburg et al. (1995) Protein Science 4, 2436 |
|
|
|
Electron density map
|
|
Where to find coordinates:
The Protein Data Bank (PDB) database
The PDB database
"The Protein Data Bank (PDB) is an archive
of experimentally determined three-dimensional structures of biological
macromolecules, serving a global community of researchers, educators, and
students. The archives contain atomic coordinates, bibliographic citations,
primary and secondary structure information, as well as crystallographic
structure factors and NMR experimental data."
As of March 4, 1998 there were 7197 released
atomic coordinate entries, distributed as 6655 proteins, peptides, and
viruses, 530 nucleic acids, 12 carbohydrates. One year later, on March
3, 1999, there are over 2200 more entries: 9419 Coordinate Entries,
8751 proteins, 656 nucleic acids and still 12 carbohydrates.
The PDB database home page is at http://www.rcsb.org/pdb
where it is easy to perform a search in the database, retrieve structure
files, verify the status of a structure which can be kept on-hold by authors
up to one year after publication.
Reference
The reference for the database is usually
given as:
Frances C. Bernstein et al., "The Protein
Data Bank: A Computer-Based Archival File for Macromolecular Structures",
Journal
of Molecular Biology, 112, 535-542, 1977.
Abola E. E. ,Bernstein F. C. ,Bryant S.
H. ,Koetzle T. F. , and Weng J. , "Protein Data Bank" in Crystallographic
Databases - Information Content, Software Systems, Scientific Applications,
eds. F. H. Allen, G. Bergerhoff, and R. Sievers, Data Commission of the
International Union of Crystallography, Bonn/Cambridge/Chester, 1987, pp.
107-132
Availability
All entries are available to the public via
the world wide web.
However some authors choose to keep their
entry on hold for as much as one year after the final acceptance. It is
possible to know which entries are on hold from the server.
PDB file names
PDB files are designated by a PDB ENTRY number
which is only 4 characters long. The PDB entries are now currently cited
by authors in their research publications.
For example the PDB entry for rhinovirus
14 is 4rhv, poliovirus type3 Sabin is 1piv,
L-Lactate dehydrogenase is 1llc and glucagon is 1gcn.
However many proteins are represented multiple
times in the database as various mutants, models, bound with various compounds,
or at various pH. For eaxmple, data for insulin in the cubic crystal form
can be found for pHs 7,9,10 and 11 with PDB entries
1aph,
1bph,
1cph
and 1dph respectively but there are still many more entries
for this compound.
Retrieving PDB files
The PDB
front page is the main source for PDB data retrieval. It is available
from within the Netscape browser or other gopher browsers.
On the mainn page enter the PDB-ID code
or use the SearchLite or SearchFileds options. For eaxmple to retrieve
a PDB file for glucagon enter the word glucagon in the Compound:
entry. You can also search by ID number or author, it may depend on the
information you already have in hand. Once you have filled one or more
of the text fields, press the Send Request button. The page will
display a list of files matching the criteria you asked. For glucagon
there is only one file 1gcn : GLUCAGON (PH 6 - PH 7 FORM).
The PDB file format
A complete guide to the PDB format can be
found at http://www.rcsb.org/pdb/
under the "General Information" entry.
The current PDB format consists of lines
of information in a file, 80 columns wide by default. Each line is called
a record. There are several different types of
records,
such as JRNL for the records listing the bibliographic references,
REMARK for authors' remarks, ATOM for the atomic coordinates etc. The records
are arranged sequentially within the file to charaterize the molecule.
Example of 2 ATOM records
ATOM 1 N HIS 1 49.668 24.248 10.436 1.00 25.00 1 1GCN 50
ATOM 2 CA HIS 1 50.197 25.578 10.784 1.00 16.00 1 1GCN 51
Each line or record starts with the record
type. The
position of characters and numbers is of the utmost importance
and cannot be changed without creating errors or crashing programs. If
you edit the PDB file with a word processor make sure you do not disrupt
the column position of characters or numbers. It is obvious that for REMARK
records
this has not the same importance.
For example the following 2 records are
not equivallent:
ATOM 1 N HIS 1 49.668 24.248 10.436 1.00 25.00 1 1GCN 50
ATOM 1 N HIS 1 49.668 24.248 10.436 1.00 25.00 1 1GCN 50
The latter record is in fact equal to:
ATOM 1 N HIS 1 9.668 4.248 0.436 0.00 0.00 1 1GCN 50
The first 3 real numbers on each line are in fact
the x,y and z coordinates of the atom in three dimensional space. The position
of this Nitrogen atom would be in a very different position with this erroneous
modification.
It is therefore very important to keep the column
arrangement unchanged. If you are editing the file with a word processor
on your desktop computer use a monospace font like courrier to display
the file.
List of records.
There are many records and subrecords
types. They are listed below, grouped by function. A pdb file containing
only
the ATOM record lines without any other information can usually
be opened without problems by visulization programs. After all this is
where the three dimensional coordinates are! Some records are very
rarely used. The records in bold in the following table are further defined
as well. Other record definitions can be found at
http://pdb.pdb.bnl.gov/Format.doc/Contents_Guide_2.html
1. Title Section
HEADER OBSLTE TITLE CAVEAT COMPND SOURCE KEYWDS EXPDTA AUTHOR REVDAT SPRSDE
JRNL REMARK REMARK 1 REMARK 2 REMARK 3 REMARK 4 - 999
2. Primary Structure Section
MODRES DBREF SEQADV SEQRES
3. Heterogen Section
HET HETNAM HETSYN FORMUL
4. Secondary Structure Section
HELIX SHEET TURN
5. Connectivity Annotation Section
SSBOND LINK HYDBND SLTBRG CISPEP
6. Miscellaneous Features Section
SITE
7. Crystallographic and Coordinate Transformation Section
CRYST1 ORIGXn SCALEn MTRIXn TVECT
8. Coordinate Section
MODEL ATOM SIGATM ANISOU SIGUIJ TER HETATM ENDMDL
9. Connectivity Section
CONECT
10. Bookkeeping Section
MASTER END
------------------------------------------------------------------------------
RECORD TYPE DESCRIPTION
------------------------------------------------------------------------------
JRNL Literature citation that defines the coordinate set.
SEQRES Primary sequence of backbone residues.
HELIX Identification of helical substructures.
SHEET Identification of sheet substructures.
TURN Identification of turns.
SSBOND Identification of disulfide bonds.
ATOM Atomic coordinate records for standard groups.
TER Chain terminator.
HETATM Atomic coordinate records for heterogens (non amino-acids)
CONECT Connectivity records.
------------------------------------------------------------------------------
Example of a PDB file
Here is a portion of the PDB file 1gcn.full describing
the structure of glucagon, a 29 amino acid peptide in a single peptide
chain in a mostly alpha helix conformation. Glucagon is a peptide hormone
involved in the cellular usage of glucose.
The file has 311 lines or records. Most
of the ATOM records are truncated here.
Note that each atom (ATOM records) for
the amino acids has a
record (assignment).
Note the function of a few records: SEQRES
provides the sequence in three letter code. HELIX is a single line and
tells which amino-acids are in an alpha-helix conformation.
HEADER HORMONE 17-OCT-77 1GCN 1GCN 3
COMPND GLUCAGON (PH 6 - PH 7 FORM) 1GCN 4
SOURCE PORCINE (SUS SCROFA) PANCREAS 1GCN 5
AUTHOR T.L.BLUNDELL,K.SASAKI,S.DOCKERILL,I.J.TICKLE 1GCN 6
REVDAT 5 30-SEP-83 1GCND 1 REVDAT 1GCND 1
REVDAT 4 31-DEC-80 1GCNC 1 REMARK 1GCND 2
REVDAT 3 22-OCT-79 1GCNB 3 ATOM 1GCND 3
REVDAT 2 29-AUG-79 1GCNA 3 CRYST1 1GCND 4
REVDAT 1 28-NOV-77 1GCN 0 1GCND 5
JRNL AUTH K.SASAKI,S.DOCKERILL,D.A.ADAMIAK,I.J.TICKLE, 1GCN 7
JRNL AUTH 2 T.BLUNDELL 1GCN 8
JRNL TITL X-RAY ANALYSIS OF GLUCAGON AND ITS RELATIONSHIP TO 1GCN 9
JRNL TITL 2 RECEPTOR BINDING 1GCN 10
JRNL REF NATURE V. 257 751 1975 1GCN 11
JRNL REFN ASTM NATUAS UK ISSN 0028-0836 006 1GCN 12
REMARK 1 1GCN 13
REMARK 1 REFERENCE 1 1GCN 14
REMARK 1 EDIT M.O.DAYHOFF 1GCN 15
REMARK 1 REF ATLAS OF PROTEIN SEQUENCE V. 5 125 1976 1GCN 16
REMARK 1 REF 2 AND STRUCTURE,SUPPLEMENT 2 1GCN 17
REMARK 1 PUBL NATIONAL BIOMEDICAL RESEARCH FOUNDATION, 1GCN 18
REMARK 1 PUBL 2 SILVER SPRING,MD. 1GCN 19
REMARK 1 REFN ISBN 0-912466-05-7 435 1GCN 20
REMARK 2 1GCN 21
REMARK 2 RESOLUTION. 3.0 ANGSTROMS. 1GCNC 1
REMARK 3 1GCN 23
REMARK 3 REFINEMENT. REALSPACE REFINEMENT AND ENERGY REFINEMENT. 1GCN 24
REMARK 4 1GCN 25
REMARK 4 THE GLUCAGON CRYSTALS ARE FORMED AT PH 9.2 AND THEN THE PH 1GCN 26
REMARK 4 IS CHANGED TO BETWEEN 6 AND 7. CRYSTALS AT BOTH PH,S HAVE 1GCN 27
REMARK 4 HIGH TEMPERATURE FACTORS, AND DATA TERMINATE AT 1GCN 28
REMARK 4 APPROXIMATELY 3 ANGSTROMS RESOLUTION. THE COORDINATES ARE 1GCN 29
REMARK 4 OBTAINED FROM THE 3 ANGSTROMS RESOLUTION ELECTRON DENSITY 1GCN 30
REMARK 4 MAP AND REFINED USING REAL SPACE REFINEMENT AGAINST 1GCN 31
REMARK 4 (2FOBS-FCALC),ALPHA CALC ELECTRON DENSITY MAPS WITH 1GCN 32
REMARK 4 GEOMETRIC RESTRAINTS, FOLLOWED BY LEVITT ENERGY 1GCN 33
REMARK 4 MINIMIZATION. NO SOLVENT CAN BE INCLUDED AT 3 ANGSTROMS. 1GCN 34
REMARK 4 WARNING - LOW RESOLUTION (3 ANGSTROMS) IMPLIES RATHER 1GCN 35
REMARK 4 INACCURATE COORDINATES AND MEANINGLESS TEMPERATURE FACTORS. 1GCN 36
REMARK 5 1GCNA 1
REMARK 5 CORRECTION. MOVE CRYST1 RECORD TO ITS PROPER POSITION. 1GCNA 2
REMARK 5 29-AUG-79. 1GCNA 3
REMARK 6 1GCNB 1
REMARK 6 CORRECTION. FIX NAMING AND HENCE ORDERING OF TWO ATOMS. 1GCNB 2
REMARK 6 22-OCT-79. 1GCNB 3
REMARK 7 1GCNC 2
REMARK 7 CORRECTION. STANDARDIZE FORMAT OF REMARK 2. 31-DEC-80. 1GCNC 3
REMARK 8 1GCND 6
REMARK 8 CORRECTION. INSERT REVDAT RECORDS. 30-SEP-83. 1GCND 7
SEQRES 1 29 HIS SER GLN GLY THR PHE THR SER ASP TYR SER LYS TYR 1GCN 37
SEQRES 2 29 LEU ASP SER ARG ARG ALA GLN ASP PHE VAL GLN TRP LEU 1GCN 38
SEQRES 3 29 MET ASN THR 1GCN 39
FTNOTE 1 1GCN 40
FTNOTE 1 RESIDUES 1 THROUGH 5 ARE RATHER DISORDERED IN THE CRYSTALS. 1GCN 41
HELIX 1 A PHE 6 LEU 26 1 1GCN 42
CRYST1 47.100 47.100 47.100 90.00 90.00 90.00 P 21 3 12 1GCNA 4
ORIGX1 .021231 0.000000 0.000000 0.000000 1GCN 43
ORIGX2 0.000000 .021231 0.000000 0.000000 1GCN 44
ORIGX3 0.000000 0.000000 .021231 0.000000 1GCN 45
SCALE1 .021231 0.000000 0.000000 0.000000 1GCN 46
SCALE2 0.000000 .021231 0.000000 0.000000 1GCN 47
SCALE3 0.000000 0.000000 .021231 0.000000 1GCN 48
ATOM 1 N HIS 1 49.668 24.248 10.436 1.00 25.00 1 1GCN 50
ATOM 2 CA HIS 1 50.197 25.578 10.784 1.00 16.00 1 1GCN 51
ATOM 3 C HIS 1 49.169 26.701 10.917 1.00 16.00 1 1GCN 52
ATOM 4 O HIS 1 48.241 26.524 11.749 1.00 16.00 1 1GCN 53
ATOM 5 CB HIS 1 51.312 26.048 9.843 1.00 16.00 1 1GCN 54
ATOM 6 CG HIS 1 50.958 26.068 8.340 1.00 16.00 1 1GCN 55
ATOM 7 ND1 HIS 1 49.636 26.144 7.860 1.00 16.00 1 1GCN 56
ATOM 8 CD2 HIS 1 51.797 26.043 7.286 1.00 16.00 1 1GCN 57
ATOM 9 CE1 HIS 1 49.691 26.152 6.454 1.00 17.00 1 1GCN 58
ATOM 10 NE2 HIS 1 51.046 26.090 6.098 1.00 17.00 1 1GCN 59
ATOM 11 N SER 2 49.788 27.850 10.784 1.00 16.00 1 1GCN 60
ATOM 12 CA SER 2 49.138 29.147 10.620 1.00 15.00 1 1GCN 61
ATOM 13 C SER 2 47.713 29.006 10.110 1.00 15.00 1 1GCN 62
ATOM 14 O SER 2 46.740 29.251 10.864 1.00 15.00 1 1GCN 63
ATOM 15 CB SER 2 49.875 29.930 9.569 1.00 16.00 1 1GCN 64
ATOM 16 OG SER 2 49.145 31.057 9.176 1.00 19.00 1 1GCN 65
/////////////////////////ATOM RECORDS TRUNCATED/////////////////////////////////
ATOM 239 N THR 29 3.391 19.940 12.762 1.00 21.00 1GCN 288
ATOM 240 CA THR 29 2.014 19.761 13.283 1.00 21.00 1GCN 289
ATOM 241 C THR 29 .826 19.943 12.332 1.00 23.00 1GCN 290
ATOM 242 O THR 29 .932 19.600 11.133 1.00 30.00 1GCN 291
ATOM 243 CB THR 29 1.845 20.667 14.505 1.00 21.00 1GCN 292
ATOM 244 OG1 THR 29 1.214 21.893 14.153 1.00 21.00 1GCN 293
ATOM 245 CG2 THR 29 3.180 20.968 15.185 1.00 21.00 1GCN 294
ATOM 246 OXT THR 29 -.317 20.109 12.824 1.00 25.00 1GCN 295
TER 247 THR 29 1GCN 296
MASTER 34 2 0 1 0 0 0 6 246 1 0 3 1GCND 8
END 1GCN 298
Coordinates and reference axes
By far the most important data are the ATOM (amino-acid
atoms), and HETATM (heterogeneous atoms of ligands)
records. The
three dimensional XYZ coordinates are the first three
real numbers
on each line. For example 49.668 24.248 10.436 for the first ATOM
record
(Nitrogen atom of Histidine amino-acid number 1).
These coordinates are in a Cartesian
coordinate system. This means that the x,y and z axes
are perpendicular to one another and their length is 1. The unit length
is 1 Å (equal to 0.1 nm [nanometer] in the international notation).
This is a prefered system of reference for most
biological users, however it is worth knowing that in some cases the frame
of reference is the length of the crystallographic "unit cell".
In this case the axes are labelled a,b and c. They
are not necessarily perpendicular to one another and do not necessarily
have the same length. If the coordinates are expressed as a function of
these axes they are usually refered to as fractional coordinates.
Most chemical databases give the coordinates in this fashion. In the PDB
formated file, he CRYST1 and SCALEn records are related to these axes but
for our purpose can be ignored.
Other formats
There are many other formats which are used in molecular
graphics!
Even the PDB format itself has some specific "expansions"
by some particular programs for their own use. However the PDB files that
you will retrieve from the PDB database will not contain any of these additions
and therefore you need not worry about this.
Other molecular graphics programs require their
own specific format for display. The program BABEL, available on all major
computer platforms (Mac, DOS and most popular Unix) reads the followinginput
formats:
Mopac Cartesian, Mopac Internal, Mopac Output,
CSD GSTAT, CSD CSSR, Free Form Fractional, Macromodel, MM2 Output, PDB
Alchemy, XYZ, Mac Molecule, Chem3D, MicroWorld, Ball and Stick, MOLIN
and writes the following output formats:
Mopac Cartesian, Mopac Internal, Gaussian Input,
IDATM, Macromodel, Mac Molecule, MM2 Input, MM2 Ouput,
PDB file,
Alchemy, XYZ, Ball and Stick, Chem3D, MicroWorld, Report of interatomic
distances,angles,and torsions.
This can give you an idea of the number of formats
"out there"! The program BABEL is available at: ftp://ccl.osc.edu/pub/chemistry/software/MAC/babel/
and has a home page at http://mercury.aichem.arizona.edu/babel.html.
Desktop computer
graphics programs
Introduction
Computer graphics programs (whether they run
on a desktop computer or on an expensive workstation) read,
interpret
and
display the PDB file (or other formats) into
graphical images
which you can manipulate in three dimensions and modify interactively on
your computer screeen.
The programs can be:
Free or Expensive
Simple or Difficult
Useful or Not so useful
Visualization or Modeling
There are many free visualization-only programs.
3D modeling is available on a limited basis in WebLabviewerLite but more
expanded in the Pro version. Most free programs are for display only.
Here are some FREE programs:
-
Long
list of FREE Software
http://klaatu.oit.umass.edu:80/microbio/rasmol/othersof.htm#viewrend
-
RasMol
(Mac, Windows, Unix). Powerful line command.
http://www.umass.edu/microbio/rasmol/
-
CHIME
Web browser plug-in. Complex menus.
http://www.mdli.com/support/chime/chimefree.htm
-
WebLab
Viewer Lite (FREE) or Pro. (Mac, Windows 95,98,NT). Beautifully renderedOpenGL
graphics.
http://www.msi.com/download/index.html
-
MOLVIEW
(Mac)- makes quicktime movies, beautiful renderings
http://www.expasy.ch/spdbv/mainpage.htmlhttp://bilbo.bio.purdue.edu/~tom/
-
SwissPDBViewer
(Mac)
http://www.expasy.ch/spdbv/mainpage.html
-
Cn3D
(See in Three-Dee) (Mac, Windows, Unix). Additional equence alignment
window.
http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml
-
kineMAGE(Mac,
Windows, Unix)
http://www.faseb.org/protein/kinemages/MageSoftware.html
Depending
on your working environment it may be worth using a program that runs on
various platforms. Rasmol is a very powerful program to explore
molecules, WebLabViewer provides superior graphics at the expense
of speed compared to rasmol. Depending on the task I use this or
that program. For example I often open the PDB files with rasmol
for a quick exploration, and then revert to a program with better graphics
for creating final images.
Rasmol reads in the PDB file directly
without
any modifications which is an adavntage. It provides a line-based and
menu-driven interface which allow scripting and the easy manipulation of
structures. This is the main program we will use.
| A new way of showing 3D structures on the world wide web
is to use 'java applets' which will display the structure right inside
your Netscape page. However there are no user controls to color or change
the display, only interactive rotation and zoom. Since we've already seen
the file for glucagon here is a small window where you can see the CA (carbone
alpha) tracing of the peptide and verify that it is in an alpha helix conformation!
|
(ALT and horizontal mouse movement will
zoom (right->left) or scale down (left->right) |
Graphical representations
The 3D coordinates of the ATOM
records
represent the position of a single point in 3D space. The molecular
graphics programs can draw a line between the various atom positions to
create a wireframe representation. If only the alpha carbones are
used then it is a
C-alpha tracing. Alternatively the program may
draw a sphere at the location of each point to represent the volume
of each atom and create a space-filling model representation. More
complex molecular graphics programs can calculate and display the continuous
molecular
surface as a continuous "skin" envelopping the molecule. Other more
cartoonish representations are the Jane Richardson's style ribbon
diagram showing the various secondary structure elements particularly well.
Here are a some examples of such representations
for the glucagon peptide:
| bonds |
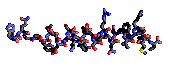 |
ribbon |
 |
| atoms |
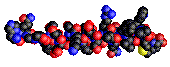 |
molecular surface |
 |
The representation of the molecule by the program will depend on what the
user
chooses amongst the program options.
The color chosen for each atom or the global colorization of
various structures or domains of the molecule will have a very important
impact on the final image for clarity and artistic value in addition to
the style chosen for the rendering.
Fortunately some programs allow mixed rendering, that is the
representation of various parts of the molecule in different styles.
In addition to the rendering of the atoms themselves, programs also
provide tools to add hydrogen bond lines or written labels at specific
location.
See examples of pre-rendered images with Rasmol at
http://heme.gsu.edu/glactone/PDB/pdb.html
RASMOL
Many of the features described above are available
in RASMOL. This program is available for all computer types and have slightly
different names on a Macintosh (RasMac), a PC (RasWin) or a unix workstation
(RasMol) but behaves essentially in the same way on all platforms. Thus,
what you learn on a Macintosh here can be used
as-is on the other
computers. This is why RASMOL has become widely used on the Internet World
Wide Web and has been chosen as the main program for this course. An offspring
of Rasmol is CHIME, a web browser plug-in with additional features. The
lack of line command makes it more complex and cumbersome to use, but can
help create beautiful "virtual museums" when all the parameters are placed
together in a web page by an instructor. For example see "The
Virtual Museum of Minerals and Molecules" (http://www.soils.wisc.edu/virtual_museum/index.html).
RasMol has been developed by Roger Sayle
(ras32425@ggr.co.uk or rasmol@dcs.ed.ac.uk) at the University of Edinburgh's
Biocomputing Research Unit and the BioMolecular Structure Department, Glaxo
Research and Development, Greenford, U.K.
The
RasMol home page is located at:
http://www.umass.edu/microbio/rasmol/
The program comes with an extensive
manual
and has an on-line
help command available. The following notes are
only a short guide to the program possibilities.
Main features
On a Macintosh, RasMol opens by double-cliking
on the RasMol program icon ( )
or a PDB file with a RasMol icon document (
)
or a PDB file with a RasMol icon document ( ).
).
This program runs on many other types
of computer, therefore, in addition to the
menu options, the program
functions with a wide array of line commands.
Upon launching, the program opens
2 windows, a graphics or canvas window which will display
the molecule and a text window for typing the additional commands
and options not found in the menu bar.
Graphics window
| The canvas window opens by default with a black background
and has two scroll bars, on the right and at the bottom. These are one
of many options to rotate the molecule interactively. The window can be
resized like any other Macintosh window.
|
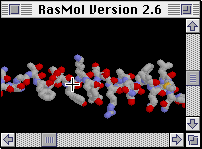 |
The mouse cursor is a cross-hair while in the canvas window, to
allow picking of atoms (for example to echo in the text window what
is the atom number).
The displayed structure can be rotated interactively with the
mouse. The manual does not give keystrokes specific for the Macintosh.
Here are summarized the movement mouse functions:
| ROTATION |
By pressing the mouse while within the canvas window the
structure can be moved in all arbirary direction ("virtual track-ball"). |
| TRANSLATION |
Pressing the OPTION
key will translate (drag) the display in the same direction as the mouse
movement. It is not necessary to depress the mouse button itself. |
| SCALING |
Pressing the SHIFT
key while moving the mouse vertically from TOP to BOTTOM zooms on the center
of the display. The BOTTOM to TOP direction would reduce the size. |
| Z-ROTATION |
Pressing BOTHSHIFT
and OPTION while moving the mouse will rotate
the molecule around the "Z axis", that is the axis which is perpendicular
to the flat screen of the computer. You are watching at the screen roughly
along this axis! |
| CLIPPING |
Pressing CONTROL
and moving the mouse will move the clipping plane if this option is enabled. |
Menu bar
The menus are located in the top menu bar
on the Macintosh, on the canvas window on other platforms. On the Macintosh
the menu bar contains the following menu and submenu items:
| File |
Edit |
Display |
Colour |
Options |
Export |
Windows |
Open...
Save As...
Page Setup...
Print...
Quit |
Undo Cut
Copy
Paste
Clear
Select All... |
Wireframe Backbone
Sticks
Spacefill
Ball & Stick
Ribbons
Strands
Cartoons |
Monochrome CPK
Shapely
Group
Chain
Temperature
Structure
User |
Slab Mode Hydrogens
Specular
Shadows
Stereo
Label |
Gif... Postcript...
PPM...
Sun Raster...
BMP...
PICT... |
Main Window Command line
|
Note: On other computers the Edit and Windows
menu items may not be present.
The Display menu provides
an easy way to change the aspect of the molecule, or portions of the molecule,
currently displayed in the canvas window. We saw examples of such representation
of the glucagon molecule earlier. The last three items (Ribbons, Strands
and Cartoons) are a variation on the ribbon diagram.
The Colour (note the British
spelling, although the program will accept the american color spelling)
menu Structure will color alpha helices, beta sheets and turns in
pink, yellow and blue respectively. It is an easy way to inspect the structure
of a new, unfamiliar molecule.
The menu has a limited number of
options. There are no submenus to choose from. Rather, most of the power
of RasMol is contained within the line commands.
Command line
The command line (text) window opens with
a statement similar to the following:
RasMol Molecular Renderer
Roger Sayle, August 1995
Version 2.6
[8bit version]
RasMol>
Interactive commands are typed at the RasMol>
prompt, but are still typed there even if the cursor is over the graphics
window.
Commands are given one at a time
on separate lines and are case INsensitive. The number of
white spaces is not important.
Rasmol recognizes a number of commands,
internal
parameters and atom expressions.
The commands give direct orders
to the program which will take immediate action. For example
color
red will color the currently selected atoms on the graphics window.
The internal parameters are
either internal default values which can be changed or, turned on or off
(boolean values: on or off, true or false). These parameters control
and alter the effect of program options and commands. For example the command
set
ssbonds backbone draw disulfide bridges between the C-alpha carbons
of cysteins in a C-alpha (RasMol Backbone) representation.
Atom expressions define groups
of atoms or subsets of a molecule in order to control how they will be
displayed. The expressions are constructed with primitive (i.e.
simple, basic) expressions and
predefined sets. Predefined sets
are abbreviations for groups of atoms for easier description. For example
hydrophobic
defines all the hydrophobic amino acids within the opened PDB file, which
is simpler than the enumeration of all of them.
Summary tables
Summary of commands/keywords currently recognised by RasMol
| backbone |
background |
cartoons |
centre |
clipboard |
colour |
connect |
cpk |
| dots |
define |
echo |
exit |
hbonds |
help |
label |
load |
| print |
quit |
renumber |
reset |
restrict |
ribbons |
rotate |
save |
| script |
select |
set |
show |
slab |
source |
spacefill |
ssbonds |
| strands |
structure |
trace |
translate |
wireframe |
write |
zap |
zoom |
List of internal parameters altered by the set
command
| ambient |
axes |
background |
bondmode |
boundbox |
display |
fontsize |
hbonds |
| hetero |
hourglass |
hydrogen |
kinemage |
menus |
mouse |
radius |
shadow |
| slabmode |
solvent |
specular |
specpower |
ssbonds |
strands |
unitcell |
vectps |
Predefined color schemes assigned by the colour
command (spelling color is accepted).
(Numbers in brakets are for Red
Green
Blue
Values
ranging from 0 to 255).
| blue |
[0,0,256] |
black |
[0,0,0] |
| cyan |
[0,255,255] |
green |
[0,255,0] |
| greenblue |
[46,139,87] |
magenta |
[255,0,255] |
| orange |
[255,165,0] |
purple |
[160,32,240] |
| red |
[255,0,0] |
redorange |
[255,69,0] |
| violet |
[238,130,238] |
white |
[255,255,255] |
| yellow |
[255,255,0] |
|
List of predefined sets
| AT |
acidic |
acyclic |
aliphatic |
alpha |
amino |
aromatic |
Backbone |
| Basic |
Bonded |
Buried |
CG |
charged |
cyclic |
cystine |
helix |
| hetero |
hydrogen |
hydrophobic |
ions |
large |
ligand |
medium |
neutral |
| nucleic |
polar |
protein |
purine |
pyrimidine |
selected |
sheet |
sidechain |
| small |
solvent |
surface |
turn |
water |
Summary classification of common amino-acids by RasMol
Predefined
set |
ALA |
ARG |
ASN |
ASP |
CYS |
GLU |
GLN |
GLY |
HIS |
ILE |
LEU |
LYS |
MET |
PHE |
PRO |
SER |
THR |
TRP |
TYR |
VAL |
|
A |
R |
N |
D |
C |
E |
Q |
G |
H |
I |
L |
K |
M |
F |
P |
S |
T |
W |
Y |
V |
| acidic |
|
|
|
* |
|
* |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| acyclic |
* |
* |
* |
* |
* |
* |
* |
* |
|
* |
* |
* |
* |
|
|
* |
* |
|
|
* |
| aliphatic |
* |
|
|
|
|
|
|
* |
|
* |
* |
|
|
|
|
|
|
|
|
* |
| aromatic |
|
|
|
|
|
|
|
|
* |
|
|
|
|
* |
|
|
|
* |
* |
|
| basic |
|
* |
|
|
|
|
|
|
* |
|
|
* |
|
|
|
|
|
|
|
|
| buried |
* |
|
|
|
* |
|
|
|
|
* |
* |
|
* |
* |
|
|
|
* |
|
* |
| charged |
|
* |
|
* |
|
* |
|
|
* |
|
|
* |
|
|
|
|
|
|
|
|
| cyclic |
|
|
|
|
|
|
|
|
* |
|
|
|
|
* |
* |
|
|
* |
* |
|
| hydrophobic |
* |
|
|
|
|
|
|
* |
|
* |
* |
|
* |
* |
* |
|
|
* |
* |
* |
| large |
|
* |
|
|
|
* |
* |
|
* |
* |
* |
* |
* |
* |
|
|
|
* |
* |
|
| medium |
|
|
* |
* |
* |
|
|
|
|
|
|
|
|
|
* |
|
* |
|
|
* |
| negative |
|
|
|
* |
|
* |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| neutral |
* |
|
* |
|
* |
|
* |
* |
* |
* |
* |
|
* |
* |
* |
* |
* |
* |
* |
* |
| polar |
|
* |
* |
* |
* |
* |
* |
|
* |
|
|
* |
|
|
|
* |
* |
|
|
|
| positive |
|
* |
|
|
|
|
|
|
* |
|
|
* |
|
|
|
|
|
|
|
|
| small |
* |
|
|
|
|
|
|
* |
|
|
|
|
|
|
|
* |
|
|
|
|
| surface |
|
* |
* |
* |
|
* |
* |
* |
* |
|
|
* |
|
|
* |
* |
* |
|
* |
|
CONCLUSION
Molecular graphics on the desktop computers
have become very powerful and it can only get better. The recent developments
combining sequence analysis and computer graphics simulations and modeling
on workstations are now ready to migrate to the desktop, or benchtop, and
will become routine companions for the analysis of protein as homology
modeling becomes more popular and more feasible with the accumulation of
new or refined molecular structures.
© 1997, 1998, 1999 Jean-Yves Sgro
last modified November 20, 1999