Ramón Roca (PDG - Centro Nacional de Biotecnología,
Madrid). ![]()
Juan Antonio García (PDG - Centro Nacional de Biotecnología,
Madrid).![]()
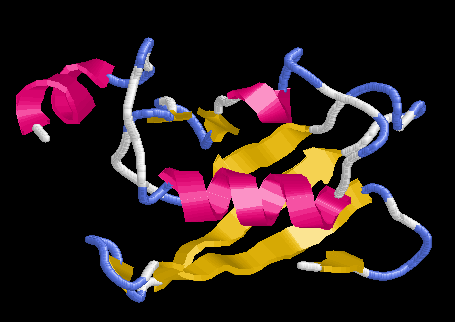
Cuando una secuencia de estructura desconocida tiene un homologo claro de estructura conocida se puede modelar basandose en esta estructura. Si la homología es alta y el alineamiento es bueno, las regiones con estructura secundaria (core de la proteina) no suelen tener problemas a la hora de modelarse, para ellas se toman las coordenadas del esqueleto y los Cb de la estructura molde. Para las regiones loop se usan distintas aproximaciones dependiendo del programa. Para las cadenas laterales se usan librerias de rotámeros y dinámica molecular o minimización de energía.
solv_prefResultado: En general cualquier valor negativo de la energía de solvatación es indicativo de estructura estable. Otra buena medida es la relación entre la energía de solvatación y el numero de átomos: si este valor es <= -0.1 se puede considerar un buen modelo.
>secuencia-problema
MSKVPRNFRL LEELEKGEKG FGPESCSYGL ADSDDITMTK WNGTILGPPH SNHENRIYSL SIDCGPNYPD SPPKVTFISK INLPCVNPTT GEVQTDFHTL RDWKRAYTME TLLLDLRKEM ATPANKKLRQ PKEGETF
Una vez que tenemos la secuencia que queremos analizar, debemos
introducirla en el programa SWISS-MODEL a través de su servidor
web, accesible en la red. En este servidor debemos introducir además
de la secuencia, nuesta dirección de "mail", nuestro nombre, y un
título para el proceso SWISS-MODEL
(First Approach mode).
AlignMaster output ============================================================ Length of target sequence: 137 residues Searching sequences of known 3D structures Found 12UCE.pdb with P(N)=5.0e-12 Found 11AYZ.pdb with P(N)=1.2e-08 Found 11AAK.pdb with P(N)=5.3e-08 Found 12AAK.pdb with P(N)=5.3e-08 Found 11UCZ.pdb with P(N)=1.1e-07 Found 12UCZ.pdb with P(N)=1.1e-07 Found 11A3S.pdb with P(N)=1.0e-05
Extracting template sequences Running pair-wise alignments with target sequence Sequence identity of templates with target: 12UCE.pdb: 22.85 % identity 11AYZ.pdb: 27.8 % identity 11AAK.pdb: 17.15 % identity 12AAK.pdb: 17.15 % identity 11UCZ.pdb: 29.6 % identity 12UCZ.pdb: 29.6 % identity 11A3S.pdb: 22.5 % identity Looking for template groups Global alignment overview: Taget Sequence: |====================================================================| 12UCE.pdb | ------------------------------------------------- 11AYZ.pdb | ------------------------------------------------ 11AAK.pdb | ----------------------------------------------------- 12AAK.pdb | ----------------------------------------------------- 11UCZ.pdb | -------------------------- 12UCZ.pdb | -------------------------- 11A3S.pdb | -------------------------------------------------- AlignMaster found 1 regions to model separately: 1: Using template(s) 11A3S.pdb 11AAK.pdb 11AYZ.pdb 11UCZ.pdb 12AAK.pdb 12UCE.pdb 12UCZ.pdb 12UCE.pdb has been rejected, too low similarity with Target sequence (22.85 % identity.) 11AAK.pdb has been rejected, too low similarity with Target sequence (17.15 % identity.) 12AAK.pdb has been rejected, too low similarity with Target sequence (17.15 % identity.) 11A3S.pdb has been rejected, too low similarity with Target sequence (22.5 % identity.)
Creating Batch files for ProMod (if any): Batch.1: residues 30 - 137 of submitted sequence. Exiting AlignMaster ProModII trace log for Batch.1 ============================================================ ProModII: Loading Template: 11AYZ.pdb ProModII: Loading Template: 11UCZ.pdb ProModII: Loading Template: 12UCZ.pdb ProModII: Loading Raw Sequence ProModII: Iterative Template Fitting ProModII: Iterative Template Fitting ProModII: Generating Structural Alignment ProModII: Aligning Raw Sequence ProModII: Refining Raw Sequence Alignment ProModII: Weighting Backbones ProModII: Averaging Sidechains ProModII: Adding Missing Sidechains ProModII: Small Ligation (C-N < 3.0A) ignored; ProModII: GROMOS will repair it at residue ASP 73 ProModII: Building CSP loop with anchor residues THR 60 and GLU 63 ProModII: Number of Ligations found: (1) ProModII: all loops are bad; continuing CSP with larger segment ProModII: Building CSP loop with anchor residues PRO 59 and GLU 63 ProModII: Number of Ligations found: (10) ProModII: ACCEPTING loop 5: clash= 1 FF= 53.8 PP=-18.41 ProModII: Dumping Preliminary Model ProModII: Dumping Sequence Alignment ProModII: Done.
Gromos96 trace log for Batch.1 ============================================================ Now running PROCS1 on file batch-procs0.dat ... Done. Now running PROCS2 on file batch-procs1.dat ... Done. Now running PROGMT on file batch-procs2.dat ... Done. Now running PROGCH on file batch-procs2.dat ... Done. Now running PROMD on file batch-progch.dat ... Done. Now running PROMD on file batch-promd0.dat ... Done. Detection of SS-Bonds within batch ...
MWVPSMYPVK PPFISINLEN FDMNTISSSL PIQEYIDSNG WIALPILHCW DPAAMNLIMV VQELMSLLHE PPQDQAPSLP PKPNTQLQQE GEYSPTAPKA QVSTPKTAIA STSTTSTGI
MLGAMAEEAVAPVAVPTTQEQPTSQPAAAQVTTVTSPSVTATAAAATAAVASPQANGNAA SPVAPASSTSRPAEELTCMWQGCSEKLPTPESLYEHVCERHVGRKSTNNLNLTCQWGSCR TTTVKRDHITSHIRVHVPLKPHKCDFCGKAFKRPQDLKKHVKTHADDSVLVRSPEPGSRN PDMMFGGNGKGYAAAHYFEPALNPVPSQGYAHGPPQYYQAHHAPQPSNPSYGNVYYALNT GPEPHQASYESKKRGYDALNEFFGDLKRRQFDPNSYAAVGQRLLSLQNLSLPVLTAAPLP EYQAMPAPVAVASGPYGGGPHPAPAYHLPPMSNVRTKNDLINIDQFLQQMQDTIYENDDN VAAAGVAQPGAHYIHNGISYRTTHSPPTQLPSAHATTQTTAGPIISNTSAHSPSSSTPAL TPPSSAQSYTSGRSPISLPSAHRVSPPHESGSSMYPRLPSATDGMTSGYTAASSAAPPST LGGIFDNDERRRYTGGTLQRARPASRAASESMDLSSDDKESGERTPKQISASLIDPALHS GSPGEDDVTRTAKAATEVAERSDVQSEWVEKVRLIEYLRNYIANRLERGEFSDDSEQEQD QEQEQDQEQEQDQEQGQDRVSRSPVSKADVDMEGVERDSLPRSPRTVPIKTDGESAEDSV MYPTLRGLDEDGDSKMPS
|
|
|
|
|
||||||
|
![]()
![]()