No todos los experimentos de DNA-arrays requieren este tratamiento a posteriori. Los DNA-arrays son sólo una técnica experimental, por lo que el diseño del experimento puede variar mucho de un caso a otro.
Por ejemplo, un experimento podría consistir en comparar el comportamiento de dos tipos de individuos, unos sanos y otros con alguna particularidad como una enfermedad, una mutación, etc. en unas condiciones determinadas para ver qué genes están implicados en dicha diferencia. En ese caso solo tendríamos que ver qué genes varían su expresión y ver en qué medida lo hacen.
Sin embargo, lo más normal es estudiar una serie de condiciones distintas (series temporales, distintas dosis de un compuesto, distintos fenotipos, etc) En este caso, sí encontramos para cada gen un patrón de expresión correspondiente a las distinas condiciones. Cuando diferentes genes presentan el mismo patron de expresión bajo las distintas condiciones experimentales estudiadas suele ser porque tienen alguna propiedad biológica en común. En este caso necesitamos conocer cuantos tipos de patrones de expresión diferentes tenemos y cuantos genes están siguiendo cada patrón.
El desarrollo clásico de un experimento de DNA-array es un estudio del nivel de expresión génica bajo distinta condiciones experimentales. Los valores de expresión génica se expresan con respecto a una condición de referencia. Dependiendo del tipo de expreimento se elige na u otra. Cuando son series temporales o de dosis, lo normales tomar la condición inicial como condición de reeferencia. En otrs casos es más complicado y suele tomarse un valor medio de expresión.
Queremos ver diferencias en la expresión de los genes entre ambas condiciones. Para eso podemos dividir los valores de la condición en estudio por los de las condiciones de referencia. Así obtendremos valores cercanos a 1 cuando el gen no haya variado su expresión. Los valores por debajo indicarán una disminución en el nivel de expresión del gen y los valores por encima, lógicamente, un aumento.
Desafortunadamente, al dividir unos valores por los otros, obtenemos valores entre 0 y 1 cuando se reprime un gen y valores entre 1 y +infinito cuando se activa. Nos gustaría tener una escala simétrica alrededor del 1, puesto que este es el valor al que corresponde un nivel de expresión igual en ambos condiciones.
Con una transformación logarítmica conseguimos este propósito. De hecho, esta es la transformación más usada. En principio cualquier otra transformación podría valer, siempre y cuando obtengamos una escala simétrica para valores de represión y de activación de los genes.
En principio, podemos suponer que genes que se comportan de la misma forma lo hacen por alguna razón biológica. Para encontrar esta función biológica, lo primero es saber cuándo podemos considerar que dos genes se comportan igual y cuándo no. Dicho de otro modo, debemos definir una distancia entre patrones.
Según la distancia que usemos para agrupar los distintos patrones de expresión, obtendremos unos grupos u otros, es decir, la distancia define la relación que buscamos entre los genes. Las distancias más usadas son la distancia euclídea y el coeficiente de correlación lineal.
Si usamos la distancia euclídea tal cual, encontraremos genes en los que su transcripción se ha activado (o reprimido) en un número de veces parecido, mientras que si usamos el coeficiente de correlación lineal, juntaremos los genes según los valores relativos y no absolutos de sus patrones de expresión. Por ejemplo:
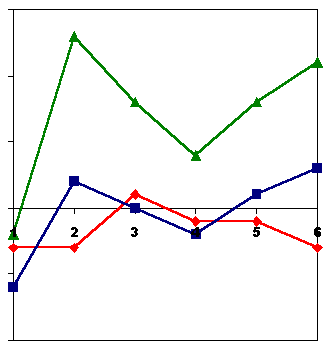
En este caso, si miramos distancia euclídea, el patrón rojo y el negro aparecerán juntos y el azul separado mientras que si medimos correlación lineal, el rojo y el azul se juntarán y el negro no.
Normalmente se usa correlación lineal, pero si se usa la distancia euclídea, se normalizan previamente los patrones de modo que estén todos comprendidos en un mismo rango de valores.
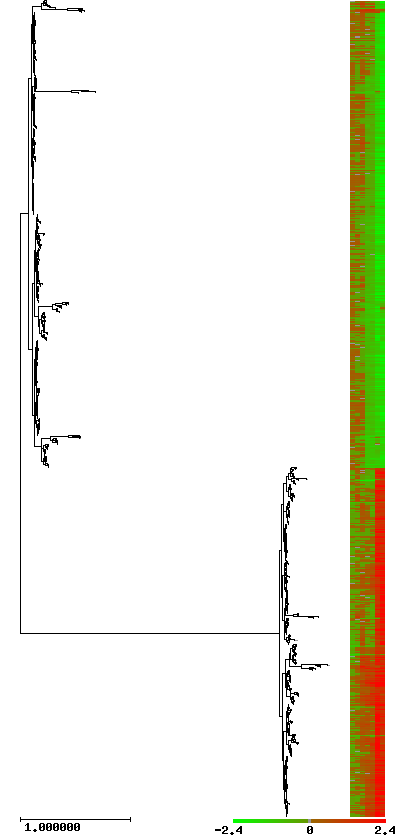
Se han usado diversos procedimientos para estudiar los clusteres de genes cuya expresion es similar. Podemos clasificarlos en jerárquicos y no jerarquicos, según nos produzcan una clasificación basada en forma de árbol binario o no den información acerca de posibles relaciones jerárquicas entre los datos. El primer algoritmo utilizado fue UPGMA, un algoritmo jerárquico aglomerativo. Los algoritmos aglomerativos son los que van juntando poco a poco los patrones más similares hasta acabar de construir el árbol completo También usaremos el SOTA (Self-Organizing Tree Algorithm), una red neuronal con topología de árbol binario. Es un método divisivo, esto es, que va separando los distintos patrones paso a paso. También existen ejemplos de algoritmos divisivos pero no jerarquicos. Quizás el mas usado de estos últimos es el SOM.
Si no disponemos de un laboratorio experimental que nos sumistre los datos, podemos usar los de otros grupos que hayan publicado sus experimentos.
Eliminar los valores en los que la expresión esté fuera de los límites de detección del método o en los que se haya detectado algún error.
Dividir los datos de expresión de las condiciones en estudio por los valores de las condiciones de referencia.
Transformar logarítmicamente los datos.
(Opcional) Eliminar los genes para los cuales las diferencias de expresión no sean significativas.
(Opcional) Normalizar los patrones de expresión.
Usaremos los procedimientos de clustering previamente descritos y compararemos los resultados

Si se dispone de datos propios, se pueden cargar directamente en el servidor: Sotarray Server [demo]
Seleccionar el experimento de Diauxic shift:
(Opcional) Se pueden comprobar el contenido de los distintos archivos e ir siguiendo los sucesivos tratamientos que han sufrido los datos
Elegir uno de los archivos (data_norm.txt, por ejemplo) y mandarlo directamente al servidor pulsando en:
Seleccionar el servidor del Sota para DNA-arrays:
|
Seleccionar la opción Unrestricted Grow.
Pulsar el botón Run.
Observar el resultado y mandarlo al TreView para visualizar el árbol:
|
Aceptar los parámetros por defecto y pulsar Run
Volver hacia atrás pulsando en:
|
Quitar la marca de la casilla Gene Names y cambiar a 1 el valor de Vertical Separation
Pulsar otra vez Run.
2a Parte:
Cerrar la ventana del árbol y volver a la del resultado del
Sotarray
Pulsar en Change Parameters para modificar los parámetros:
|
Seleccionar la opción Variablity Threshold (%) y poner el valor de 90 (por defecto).
Volver a pulsar el botón Run.
Observar el resultado y mandarlo tanto al TreeView como al SotaTree para visualizar el árbol:
|
|
Comparar y comentar los resultados
Si se dispone de datos propios, se pueden cargar directamente en el servidor: Cluster Server [demo]
Desde la página de data sets, elegir uno de los experimentos.
Elegir uno de los archivos y cargarlo directamente al servidor pulsando en:
Mandarlo al Cluster:
|
Aceptar los valores por defecto (UPGMA y correlation) y pulsar Run
Mandar el resultado al TreeView para visualizarlo:
|
Quitar la opción de Gene Names cambiar el valor de Vertical Separation a 1 y pulsar Run
Si se dispone de datos propios, se pueden cargar directamente en el servidor: SOM Server [demo]
Desde la página de data sets, elegir uno de los experimentos.
Elegir uno de los archivos y cargarlo directamente al servidor pulsando en:
Mandarlo al SOM:
|
Aceptar los valores por defecto y pulsar Run
Comparar y comentar los resultados
Abrir la página de data sets pulsando aquí:
Si se dispone de datos propios, se pueden cargar directamente en el servidor:
Si todavía queda tiempo, elegir un cluster de los obtenidos anteriormente, que contenga entre 5 y 10 genes más o menos y buscar información sobre la función de cada gen.
Se puede encontrar mucha información en la base de datos Swiss-Prot:
Intentar asignar una función global al cluster.




